
Lay Summary
The attachment of sugars chains (glycans) to proteins is known as protein glycosylation. Protein glycosylation can be divided into two main categories, N-linked and O-linked protein glycosylation, depending on how sugars are added to proteins1. N-linked protein glycosylation (or N-glycosylation) is the process by which glycans are attached to the amino acid asparagine in proteins. When a glycan is attached to a protein in this way, the product is called an N-linked glycoprotein. Any mutation that prevents the proper formation of N-linked glycoproteins is known as a disorder of N-linked protein glycosylation2. Disorders of N-linked protein glycosylation are the most common type of congenital disorder of glycosylation (CDG), and 36 known N-linked CDG have been identified to date3. Due to the important roles that N-glycosylated proteins play in many different biological pathways, multiple systems are usually affected in people with N-linked CDG. Although symptoms, clinical manifestations, severity and age of onset vary between different N-linked CDG, some common signs include developmental delays, neurologic disease, failure to thrive, poor muscle tone, abnormal facial features, blood clotting and hormonal abnormalities; issues with the heart, liver, and kidneys are also common2,4. The first step in diagnosis of N-linked CDG involves blood tests on a specific protein, transferrin, but definitive diagnosis typically requires genetic testing. A few N-linked CDG are treatable, and early diagnosis is crucial to inform care for affected individuals5,6.
N-Linked Protein Glycosylation
N-linked protein glycosylation (or N-glycosylation) involves the attachment of an 14-sugar glycan to the nitrogen atom of asparagine (Asn) residues on a protein as it is being synthesized in the endoplasmic reticulum (ER) lumen7,8. N-glycoproteins play a key role in multiple biological pathways, and N-linked glycans can affect protein localization, stability, solubility, activity, and interactions8,9.
The N-glycosylation pathway can be divided into the following steps:
- Lipid-linked oligosaccharide (LLO) biosynthesis
- N-glycosylation and protein folding
- N-glycan remodeling
The first step in the N-glycosylation pathway is the synthesis of the oligosaccharide precursor that will be attached to the asparagine residue of a protein. The oligosaccharide is assembled on dolichol, a lipid anchored in the ER membrane (Figure 1)8. This molecule, comprised of the oligosaccharide and the lipid dolichol, is referred to as the lipid-linked oligosaccharide (LLO). Once assembled, the oligosaccharide is transferred from the LLO onto a protein, becoming an N-glycan. Then, the N-glycoprotein is folded and the glycan undergoes additional processing in the ER. The N-glycoprotein is transported to the Golgi apparatus where the N-glycan is further remodeled, yielding a complex and mature N-glycoprotein (Figure 2). The mature N-glycoprotein is then transported to the cell surface, where it is either secreted or incorporated into the plasma membrane.
Lipid-Linked Oligosaccharide (LLO) Biosynthesis
N-linked glycosylation begins with the synthesis of the LLO. The 14-sugar oligosaccharide, also referred to as the N-glycan precursor oligosaccharide, is comprised of two glucose (Glc3), nine mannose (Man9) and two N-acetylglucosamine (GlcNAc2) molecules. This oligosaccharide, Glc3Man9GlcNAc2, is constructed in a stepwise fashion on dolichol, a lipid molecule that is anchored to the ER membrane; this sugar-lipid hybrid is also known as a lipid linked oligosaccharide (LLO). Each sugar is added to the growing oligosaccharide chain on the LLO by the action of glycosyltransferases, enzymes that catalyze the transfer of sugars from a glycosyl donor to molecules which “accept” sugars. In the early steps of LLO biosynthesis, highly energetic nucleotide sugars (e.g. GDP-mannose and UDP-GlcNAc) in the cytoplasm are the glycosyl donors, but later steps of LLO synthesis occur in the ER lumen and use sugars linked to other dolichol molecules8. The synthesis of the N-glycan precursor oligosaccharide is completed inside the ER so that it is near the newly formed proteins that will be targeted for glycosylation.
LLO biosynthesis starts with the addition of two N-acetylglucosamines (GlcNAc2) to dolichol phosphate, with a pyrophosphate (PP) bond being formed between the sugar and the lipid (Figure 1)7,8. Five mannoses (Man5) are added, forming Man5GlcNAc2-PP-Dol, which is then transferred from the cytoplasmic side of the ER membrane to the ER lumen. An additional four mannoses and three glucoses are added to form the LLO, Dol-PP-GlcNAc2Man9Glc3. Inside the ER lumen, glucose and mannose sugars are linked to other phosphorylated dolichol molecules, and the sugar donors are accordingly Dol-P-mannose (Dol-P-Man) and Dol-P-glucose (Dol-P-Glc)7,8.
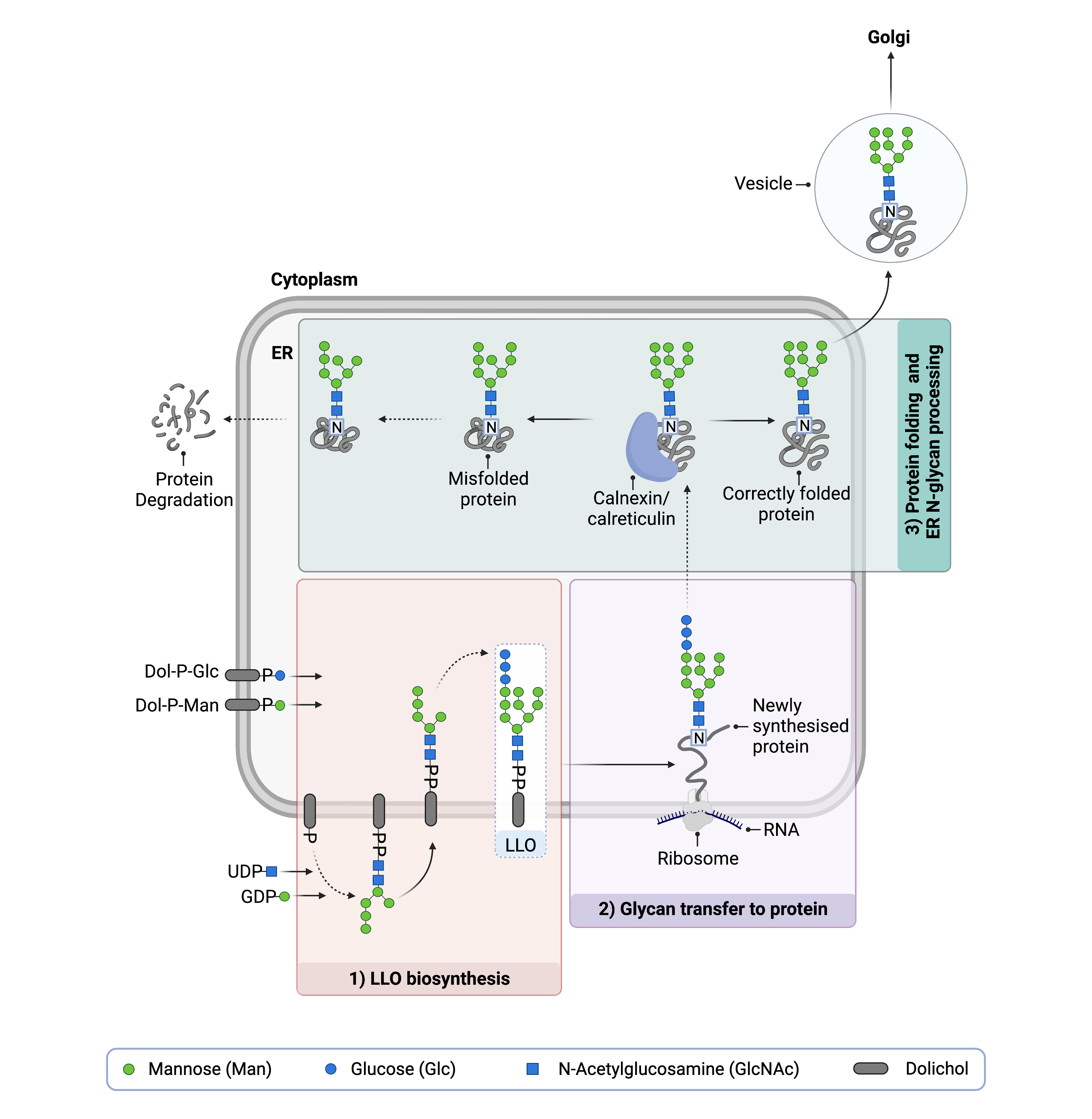
Figure 1. Lipid-linked oligosaccharide (LLO) biosynthesis and N-glycosylation.
Synthesis of the LLO begins on the cytoplasmic side of the ER; GlcNAc and mannose residues are transferred from nucleotide sugars (UDP-GlcNAc; GDP-Man) onto Dol-P. The intermediate structure is flipped into the ER lumen where mannose and glucose sugars are transferred from dolichol-linked sugars (Dol-P-Man; Dol-P-Glc) onto the growing oligosaccharide. The complete oligosaccharide, GlcNAc2Man9Glc3, is transferred onto an asparagine amino acid of a newly-synthesized protein. The N-glycoprotein undergoes folding and exits the ER.
N-Glycosylation and Protein Folding
Once assembled on the LLO, the N-glycan precursor oligosaccharide Glc3Man9GlcNAc2 is transferred to asparagine residues on newly synthesized proteins via the oligosaccharyltransferase (OST) enzyme. This process is called N-glycosylation and the attachment occurs between the nitrogen atom of the asparagine and the innermost GlcNAc in the glycan chain. The asparagine must be located in regions of the protein where the sequence of amino acids is asparagine-X-serine/threonine, also called an Asn-X-Ser/Thr consensus sequence (where X is any amino acid except proline)10.
After attachment of the glycan to the polypeptide, the three glucose residues are sequentially removed from the glycan chain by glycosidases, and the resultant N-glycoprotein undergoes additional folding with the help of chaperones8. If the protein is folded correctly, removal of the terminal glucose is a signal for the N-glycoprotein to be transported to the Golgi apparatus. However, if the protein is not folded correctly, the enzyme UDP-glucose:glycoprotein glucosyltransferase (UGGT) will re-add a glucose molecule to the glycan chain11. Next, two chaperones, calreticulin and calnexin, bind to the misfolded-protein and prevent it from leaving the ER12–14. Misfolded N-glycoproteins retained in this manner will eventually be marked for degradation.
N-Glycan Remodeling
Properly folded N-glycoproteins are transported to the Golgi. Then, the N-glycoproteins move through the Golgi and have different sugars added or removed from their glycan chain (Figure 2). This process is known as glycan remodeling, and it results in the formation of three distinct classes of N-glycans8,15:
- High-mannose - glycans that consist of many mannose sugars
- Complex - glycans that contain many different types of sugars
- Hybrid - glycans where one branch is high-mannose and the other branch is complex
After N-glycoproteins pass through the Golgi, they are transported by vesicles, to the cell surface where they are either secreted or incorporated into the plasma membrane.
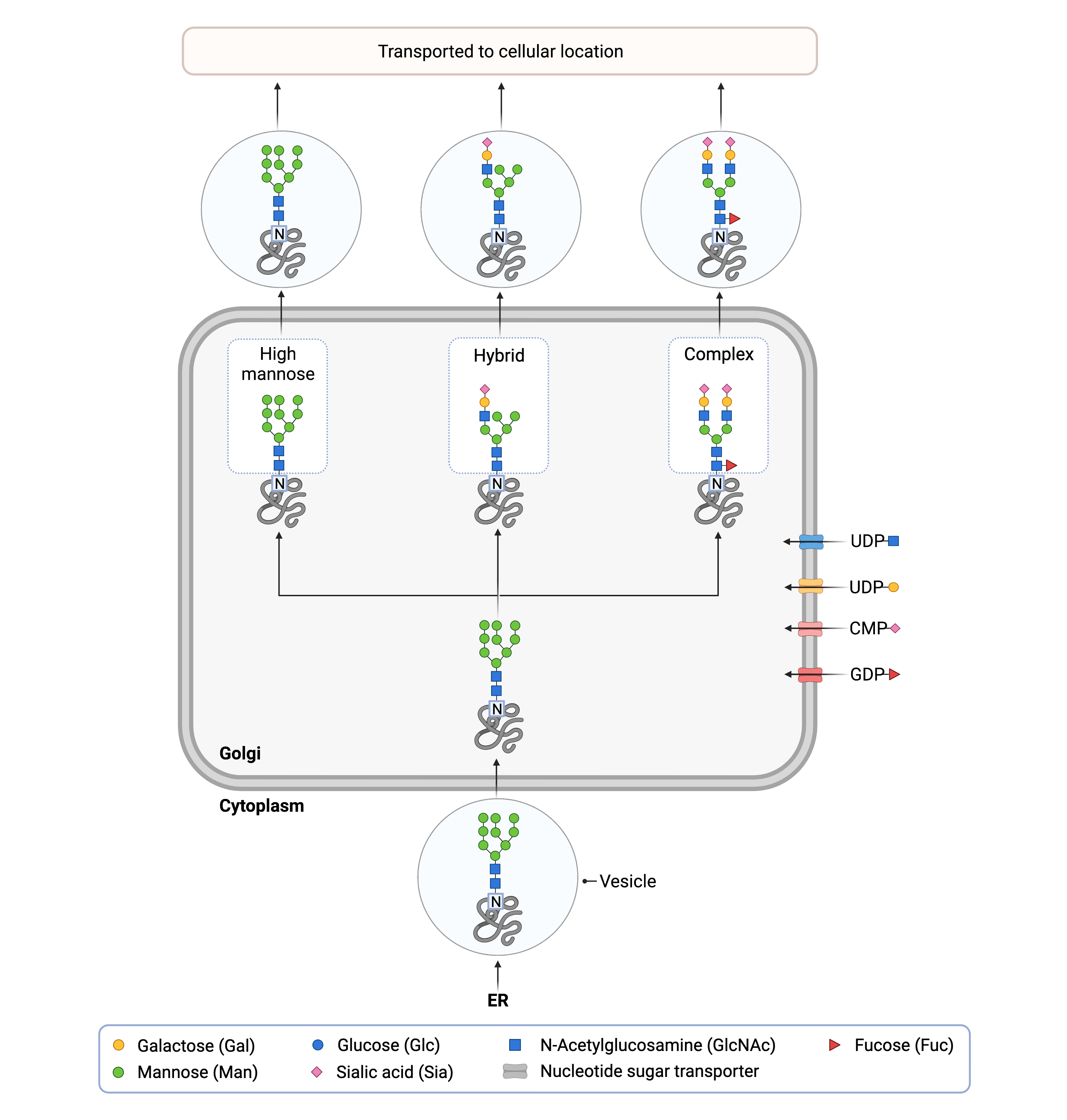
Figure 2. N-glycan remodeling in the Golgi.
An N-glycosylated protein is delivered from the ER to the Golgi by vesicles. Once inside the Golgi, sugars can be added or removed from the N-glycan, forming high-mannose, hybrid or complex glycans.
Disorders of N-Linked Protein Glycosylation
Disorders of N-linked glycosylation are the most common type of CDG, with 32 N-linked CDG currently known3. With the exception of GANAB-CDG and PRKCSH-CDG, N-linked CDG are inherited in an autosomal recessive fashion, with affected individuals receiving one defective allele from each parent2. Both GANAB-CDG and PRKCSH-CDG are inherited in an autosomal dominant fashion, meaning that affected individuals only need to inherit a single defective allele from either parent16,17.
N-linked CDG can be caused any mutation that prevents the normal formation of N-glycoproteins, including defects in synthesis of monosaccharides used in N-glycosylation, LLO synthesis, glycan transfer by the OST enzyme, and subsequent glycan remodeling (Figure 3) 18. Mutations that decrease the availability of sugar substrates can also cause N-linked CDG19. Historically, CDG were categorized as Type I CDG or Type II CDG, depending on whether the glycosylation defect occurred before or after the transfer of the oligosaccharide to the protein, originating in the ER/cytoplasm or Golgi apparatus, respectively20,21. However, this classification system only applies to N-linked CDG.
Disorders of N-linked protein glycosylation that have been identified to date include:
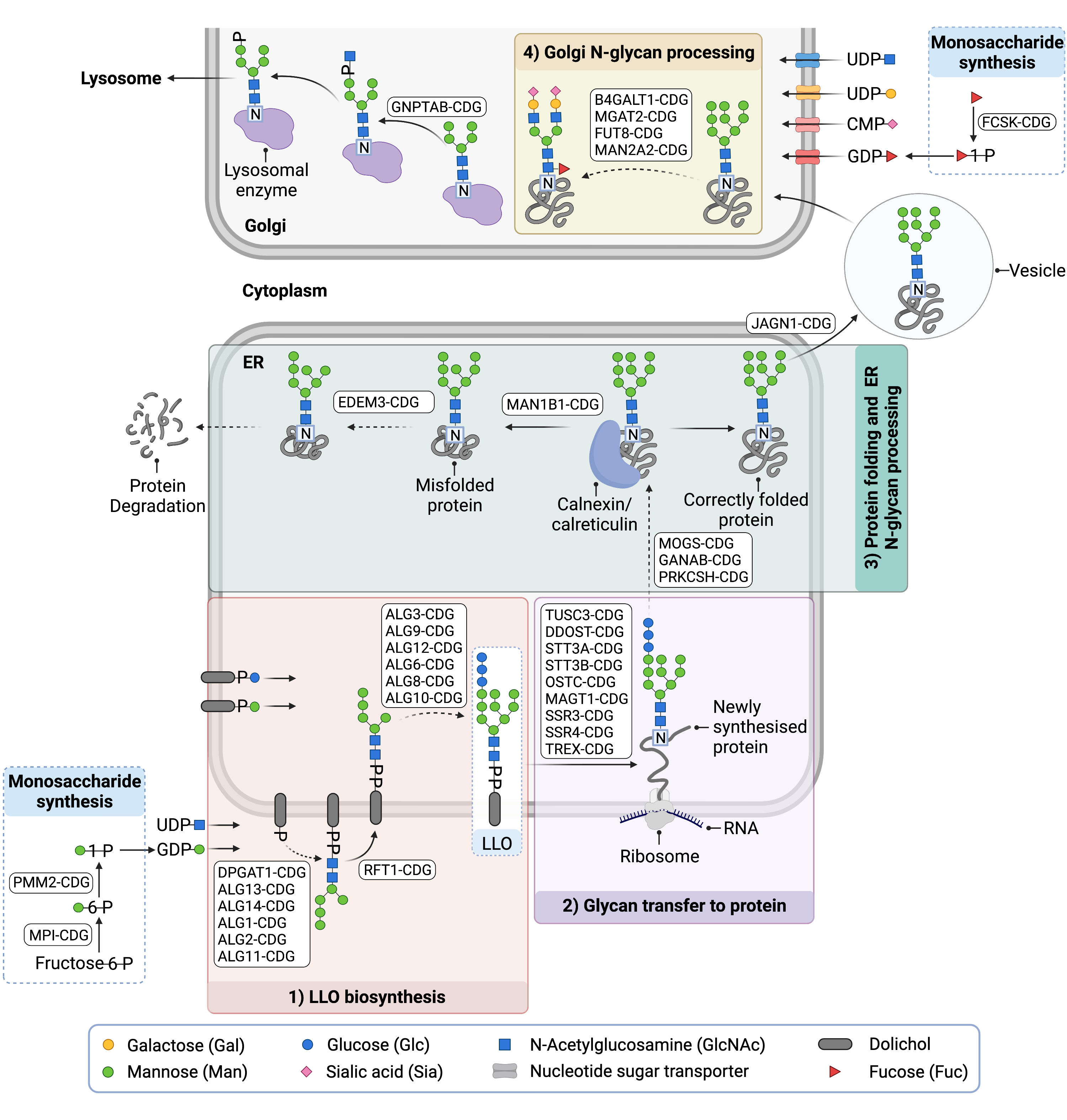
Figure 3. Overview of CDG involving N-glycosylation defects.
N-linked CDG are caused by defects in the synthesis of monosaccharides used in N-glycosylation, LLO biosynthesis, glycan transfer to a protein, and subsequent glycan remodeling in the ER or Golgi.
Diagnosis
Like all CDG, disorders of N-linked glycosylation vary widely in their clinical presentation but typically manifest symptoms in multiple systems. Common presentations include neurological abnormalities and developmental/cognitive delay, failure to thrive, poor muscle tone (hypotonia), liver disease, and clotting abnormalities. Cardiac, ophthalmological, dermatological, and renal abnormalities may also be present.
Diagnosis of N-linked CDG is achieved by conducting a series of tests which can help narrow down which part of the glycosylation pathway is affected and potentially identify individual CDG. A flowchart for the diagnosis of N-linked CDG is presented in Figure 41. The first step is typically analysis of the glycans (glycoforms) present on serum transferrin, an N-glycoprotein that is important for iron transport in the blood, using either transferrin isoelectric focusing (TIEF) or mass spectrometry22. The results from these tests may indicate whether the glycosylation defect arises in the ER, cytoplasm, or in the Golgi, and are used to inform subsequent testing. Depending on the initial findings, next steps may include analysis of specific enzymes involved in N-glycosylation and analysis of the LLO. Genetic testing is often necessary for definitive diagnosis.2,4,23,24.
Transferrin Analysis
Analysis of serum transferrin, also referred to as transferrin isoform analysis or carbohydrate deficient transferrin (CDT) analysis, is the first-line test for diagnosing N-linked CDG. Serum transferrin normally has two N-glycans with a total of four sialic acid sugars referred to as tetrasialotransferrin (Figure 4) 22.
Figure 4: Serum Transferrin Isoforms.
Transferrin contains two N-glycans and has different isoforms (glycoforms) depending on the number of sialic acid residues present on the glycans. Transferrin isoforms are named according to the number of sialic acid residues present. Tetrasialo-transferrin is the most abundant isoform in healthy individuals.
In the case of N-linked CDG, transferrin usually has altered or reduced glycosylation, with changes to the number of sialic acids in the N-glycans or chains missing entirely (Figure 5). There are several methods to analyze transferrin, but differences in the abundance of transferrin isoforms is often measured using TIEF or mass spectrometry.
- Type I pattern – characterized by low overall levels of glycosylation, represented by elevated levels of asialo- (no sialic acids) or monosialotransferrin (one sialic acid) isoforms, instead of the normal tetrasialotransferrin (Figure 5)22.
- Type II pattern – characterized by elevated levels of trisialo- (three sialic acids), disialo-, monosialo- and/or asialotransferrin.
- Mixed Type I/II pattern: a mixed transferrin profile of type I and type II patterns, characterized by elevated levels of asialo-,monosialo-, disialo-, and trisialotransferrin.
Figure 5: Example of transferrin isoelectric focusing (TIEF) results for healthy and N-linked CDG samples.
Compared to healthy samples (left), CDG Type I CDG (middle) are characterized by increased levels of asialo- and diasialo-transferrin and Type II CDG (right) are characterized by increased levels of asialo-, monosialo-, disialo-, and/or trisialo-transferrin, as indicated by darker bands on the gel.
Total N-Glycan Analysis
Several N-linked CDG types are better diagnosed by looking at all N-glycans present in a patient sample (e.g. serum, plasma, or urine) in order to identify abnormal N-glycan patterns25. This is often achieved using mass spectrometry.
Enzyme Analysis
Diagnosis of some CDG can be confirmed by testing the activity of the affected enzymes. In particular, if a Type I pattern is detected by TIEF, direct testing of phosphomannomutase 2 (PMM2) or phosphomannose isomerase (MPI) in patient derived cells can be used to quickly identify the CDG caused by defects in these enzymes1,2.
LLO Analysis
Patient fibroblast cells obtained from a skin biopsy may be used to study the structure of the LLO26–28. An accumulation of an unusual or incomplete LLO structure is indicative of a Type I CDG.
Genetic Testing
Genetic testing is often required to definitively diagnose N-linked CDG and may include targeted sequencing of specific genes, use of CDG gene panels that test for known Type I or Type II CDG, whole genome sequencing (WGS), and the sequencing of all expressed genes (whole exome sequencing, WES)2,4,23,24
References
- Péanne, R. et al. Congenital disorders of glycosylation (CDG): Quo vadis? Eur. J. Med. Genet. 61, 643–663 (2018).
- Chang, I. J., He, M. & Lam, C. T. Congenital disorders of glycosylation. Ann. Transl. Med. 6, 1–13 (2018).
- Ondruskova, N., Cechova, A., Hansikova, H., Honzik, T. & Jaeken, J. Congenital disorders of glycosylation: Still “hot” in 2020. Biochim. Biophys. Acta - Gen. Subj. 1865, (2021).
- Francisco, R. et al. The challenge of CDG diagnosis. Mol. Genet. Metab. 126, 1–5 (2019).
- Verheijen, J., Tahata, S., Kozicz, T., Witters, P. & Morava, E. Therapeutic approaches in Congenital Disorders of Glycosylation (CDG) involving N-linked glycosylation: an update. Genet. Med. 22, 268–279 (2020).
- Brasil, S. et al. CDG therapies: From bench to bedside. Int. J. Mol. Sci. 19, 1–48 (2018).
- Li, S. T. et al. Reconstitution of the lipid-linked oligosaccharide pathway for assembly of high-mannose N-glycans. Nat. Commun. 10, 1–11 (2019).
- Taylor, M. E. & Drickamer, K. Introduction to Glycobiology. (OUP Oxford, 2011).
- Imperiali, B. & O’Connor, S. E. Effect of N-linked glycosylatian on glycopeptide and glycoprotein structure. Curr. Opin. Chem. Biol. 3, 643–649 (1999).
- Mellquist, J. L., Kasturi, L., Spitalnik, S. L. & Shakin-Eshleman, S. H. The amino acid following an Asn-X-Ser/Thr sequon is an important determinant of N-linked core glycosylation efficiency. Biochemistry 37, 6833–6837 (1998).
- Dejgaard, S., Nicolay, J., Taheri, M., Thomas, D. Y. & Bergeron, J. J. M. The ER glycoprotein quality control system. Curr. Issues Mol. Biol. 6, 29–42 (2004).
- Ganan, S., Cazzulo, J. J. & Parodi, A. J. A major proportion of N-glycoproteins are transiently glucosylated in the endoplasmic reticulum. Biochemistry 30, 3098–3104 (2002).
- Gelebart, P., Opas, M. & Michalak, M. Calreticulin, a Ca2+-binding chaperone of the endoplasmic reticulum. Int. J. Biochem. Cell Biol. 37, 260–266 (2005).
- Hammond, C., Braakman, I. & Helenius, A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc. Natl. Acad. Sci. 91, 913–917 (1994).
- Higel, F., Seidl, A., Sörgel, F. & Friess, W. N-glycosylation heterogeneity and the influence on structure, function and pharmacokinetics of monoclonal antibodies and Fc fusion proteins. Eur. J. Pharm. Biopharm. 100, 94–100 (2016).
- Porath, B. et al. Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am. J. Hum. Genet. 98, 1193–1207 (2016).
- Drenth, J. P. H., te Morsche, R. H. M., Smink, R., Bonifacino, J. S. & Jansen, J. B. M. J. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat. Genet. 2003 333 33, 345–347 (2003).
- Witters, P. & Morava, E. Congenital Disorders of Glycosylation (CDG): Review. eLS 1–6 (2016) doi:10.1002/9780470015902.A0026783.
- Witters, P., Cassiman, D. & Morava, E. Nutritional therapies in congenital disorders of glycosylation (CDG). Nutrients 9, 1–10 (2017).
- Patterson, M. C. Metabolic mimics: The disorders of N-linked glycosylation. Semin. Pediatr. Neurol. 12, 144–151 (2005).
- Jaeken, J., Hennet, T., Matthijs, G. & Freeze, H. H. CDG nomenclature: Time for a change! Biochim. Biophys. Acta - Mol. Basis Dis. 1792, 825–826 (2009).
- Wopereis, S. et al. Patients with unsolved congenital disorders of glycosylation type II can be subdivided in six distinct biochemical groups. Glycobiology 15, 1312–1319 (2005).
- Timal, S. et al. Gene identification in the congenital disorders of glycosylation type i by whole-exome sequencing. Hum. Mol. Genet. 21, 4151–4161 (2012).
- Bruneel, A., Cholet, S., Tran, N. T., Mai, T. D. & Fenaille, F. CDG biochemical screening: Where do we stand? Biochim. Biophys. Acta - Gen. Subj. 1864, (2020).
- Van Scherpenzeel, M., Steenbergen, G., Morava, E., Wevers, R. A. & Lefeber, D. J. High-resolution mass spectrometry glycoprofiling of intact transferrin for diagnosis and subtype identification in the congenital disorders of glycosylation. Transl. Res. 166, 639-649.e1 (2015).
- Imbach, T. et al. Deficiency of dolichol-phosphate-mannose synthase-1 causes congenital disorder of glycosylation type Ie. J. Clin. Invest. 105, 233 (2000).
- García-Silva, M. T. et al. Congenital disorder of glycosylation (CDG) type Ie. A new patient. J. Inherit. Metab. Dis. 27, 591–600 (2004).
- Denecke, J. et al. Congenital disorder of glycosylation type Id: Clinical phenotype, molecular analysis, prenatal diagnosis, and glycosylation of fetal proteins. Pediatr. Res. 58, 248–253 (2005).