
This page is intended for healthcare professionals.
Overview
Congenital Disorders of Glycosylation (CDG) are a large group (>170 types) of genetic, multi-system and heterogeneous disorders that arise from aberrant glycosylation (synthesis and attachment of glycans), leading to abnormal glycan structures on proteins and lipids1. These disorders are classified based on the affected gene and grouped by the affected glycosylation pathway2. Glycosylation is where glycan structures modify proteins or lipids through N-, O- or C-glycosylation (which refer to a glycan linkage forming through a nitrogen, oxygen or carbon atom, respectively) and is required for normal functioning of a wide variety of biological processes, such as immunity and development3. With glycoproteins and glycolipids found abundantly throughout the body, dysfunctional glycosylation can lead to multisystem abnormalities and mild to severe disease4. The clinical manifestation of CDG varies between types and individuals, but many affected individuals present neurological abnormalities5. In addition to the broad clinical spectrum of CDG, diagnosis can be further complicated by clinical features that overlap with other diseases6. Consequently, CDG diagnosis often relies on a combination of clinical assessment, glycan analysis, enzyme analysis and DNA sequencing7. Some CDG types are treatable, often with dietary supplementation, and a number of therapies are currently under investigation, some of which are in clinical trials.
Epidemiology
The incidence and prevalence of CDG are not well established but are thought to remain largely underdiagnosed1. Individuals with CDG have been reported worldwide from most ethnic backgrounds.
The prevalence of CDG in European populations has been estimated to range from 1/10,000 to 0.1–0.5/100,0001. There are limited reports of CDG prevalence within specific countries or populations. The prevalence of CDG in Poland is estimated to be 0.1/100,0008. The estimated prevalence of CDG in the Saudi population is 1.4/100,0009. And the prevalence of CDG in the African American population is estimated to be 1/10,00010.
Genetics
The majority of CDG have autosomal recessive inheritance.
Some CDG may have autosomal dominant inheritance: GANAB-, PRKCSH-, EXT1-, EXT2-, POFUT1-, POGLUT1-CDG and SEC63-CDG; or X-linked inheritance: ALG13-, SSR4-, PIGA, SLC9A7-, SLC35A2-, ATP6AP1-, ATP6AP2, OGT- and VMA21-CDG.
De novo variants can be determined by parental testing5,10.
Classification
CDG are termed by the affected gene (non-italicized), followed by “-CDG” (such as PMM2-CDG)11. CDG can be grouped into disorders of N-linked protein glycosylation; disorders of O-linked protein glycosylation; disorders of lipid glycosylation and GPI biosynthesis; disorders of multiple glycosylation pathways; and disorders of deglycosylation (known as congenital disorders of deglycosylation; CDDG).
CDG were traditionally classified based on transferrin isoform analysis (see “Diagnosis” section for further details). Briefly, transferrin isoform analysis can generate two patterns; a type I pattern or a type II pattern, and CDG were classified as CDG-I and CDG-II, respectively, depending on the pattern displayed during transferrin isoform analysis. Within these groups, each new disorder identified was designated a letter, such as CDG-Ia. However, CDG nomenclature has been updated to the classification system listed above (i.e., “GENE-CDG”) as transferrin isoform analysis can be inconsistent within disorders and individuals, and not all CDG present abnormal transferrin11.
Symptoms
CDG are often multisystem disorders that present broad clinical features (within and among disorders) that are often associated with neurological conditions5. Symptoms can manifest from birth and range in severity, from mild to life-threatening4. The full clinical presentation of a given CDG may be unknown due to limited reported cases, but can be:
- Cardiological – cardiomyopathy, pericardial effusion and congenital heart defects
- Dermatological – ichthyosis, dyspigmentation, loose skin and lipodystrophy
- Endocrine – tumoral calcinosis, advanced bone age and hypogonadotropic hypogonadism
- Gastroenterological – failure to thrive, gastro-esophageal reflux, protein losing enteropathy and isolated polycystic liver disease
- Hematological – thrombosis, coagulopathy, isolated leukocyte adhesion deficiency and immunodeficiency
- Hepatic – hepatomegaly and fibrosis
- Musculoskeletal – skeletal dysplasia, short stature and hypermobility
- Neurological – developmental delay, hypotonia, seizures and intellectual disability
- Ophthalmologic – retinitis pigmentosa, alacrima and strabismus
- Renal – proteinuria, obstructive uropathy and cysts10
Some disorders can also be associated with particular conditions or syndromes, such as:
- Nonsyndromic retinitis pigmentosa in DHDDS-CDG
- Tumoral calcinosis in GALNT3-CDG
- Schneckenbecken dysplasia in SLC35D1-CDG5.
Diagnosis
As clinical presentation can be broad and affect multiple systems, a series of tests are often required for CDG diagnosis, such as glycan analysis (often through serum isoform analysis, mass spectrometry, capillary zone electrophoresis and HPLC), enzyme analysis and/or molecular sequencing (Figure 1)7. See the Mayo Clinic CDG Screening Algorithm here
Serum transferrin isoform analysis and apolipoprotein CIII isoform analysis are typically among the first CDG screening tests, which can determine disorders in N-glycosylation (transferrin), O-glycosylation (apolipoprotein C-III) or multiple glycosylation pathways (both). These tests identify aberrant glycosylation patterns on proteins that can be diagnostic for a group or type of CDG12.
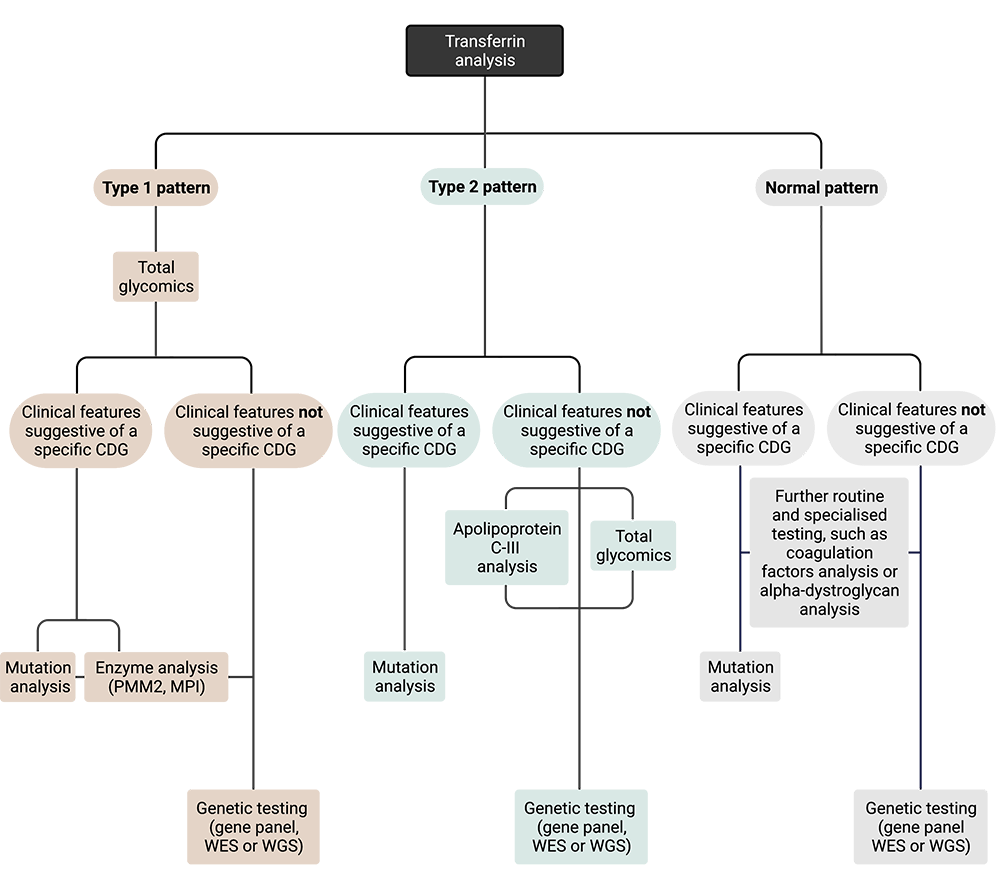
Figure 1. Overview of diagnostic workflow for assessing individuals with suspected CDG.
Diagnosis typically begins with serum transferrin isoform analysis. If a type I pattern is observed, follow up testing may include total glycomics and/or enzyme analysis if specific CDG type is suspected. If a type II pattern is observed, follow up testing typically includes analysis of apolipoprotein C-III. Transferrin and apolipoprotein CIII analysis results may be normal for a number of CDG types. Genetic testing is necessary for a definitive diagnosis of CDG and the specific CDG type.
Transferrin Analysis
Transferrin is a plasma protein that has two N-glycosylation sites, existing as different glycoforms with tetrasialo-transferrin (4 sialic acids) as the most abundant glycoform. Abnormalities in transferrin glycosylation can alter the abundance of these glycoforms, generating transferrin patterns that can be used in CDG diagnosis13:
- Type I pattern: decreased tetrasialo-transferrin and increased asialo-/disialo-transferrin glycoforms14, which is associated with defects in the cytosol or endoplasmic reticulum glycosylation defects15,16.
- Type II pattern: increased asialo-/monosialo-/disialo-/trisialo-transferrin glycoforms14, which can be associated with Golgi glycosylation defects15,16.
Transferrin isoform analysis (also known as carbohydrate deficient transferrin (CDT) analysis) was the first screening test to emerge for CDG, and these patterns were originally used to classify CDG before inconsistencies were identifies—leading to an updated CDG classification system (as detailed above)11. Analysis of transferrin isoforms is often carried out through transferrin isoelectric focusing (TIEF), mass spectrometry, or high-performance liquid chromatography (HPLC). With regards to CDG diagnosis, transferrin isoform analysis can present false negatives as individuals may present normal patterns in early life (0–3 months old)17. Transferrin patterns can also be inconsistent between affected individuals and CDG can present normal glycosylation patterns (depending on the affected gene) and can only detect N-linked CDG11. In addition, abnormal transferrin glycosylation can also be observed in other conditions, such as galactosemia and infection, leading to false positives10. However, further tests can be performed based on serum isoform analysis. If transferrin isoform analysis is suggestive of a particular defective enzyme, such as phosphomannomutase 2 (PMM2), enzyme analysis can be performed from patient fibroblast or leukocyte samples18. Alternatively, or in addition, further glycan analysis or genetic sequencing can be performed10.
Apolipoprotein CIII Analysis
Apolipoprotein C-III isoform analysis can identify defects in O-glycan biosynthesis, as apolipoprotein C-III is glycosylated with a single core 1 mucin type O-glycan. These glycosylation defects can be detected by increased abundance of asialo-apolipoprotein C-III and/or increased abundance of monosialo-apolipoprotein C-III glycoforms12.
Total Glycan Analysis
In addition to transferrin and apolipoprotein C-III glycan analysis, total N- and/or O-glycans can be assessed in a sample, using mass spectrometry to detect any abnormal glycans. Patient samples are collected and enzymatically or chemically treated to cleave glycans from the proteins. These samples are typically blood, but can also include urine, cerebrospinal fluid cells, or isolated proteins.
Molecular Testing
CDG are confirmed by molecular testing, which may involve single-gene, gene-panel, whole-exome or whole-genome sequencing. Single-gene sequencing is useful in cases where initial clinical assessment and/or glycan analysis are suggestive of a particular pathogenic variant and more extensive sequencing strategies can be used when the genetic defect is unknown19.
Differential Diagnosis
As CDG often present as multisystem disorders, affected individuals may be incorrectly diagnosed with other disorders, such as mitochondrial, peroxisomal or lysosomal storage diseases. Many of these disorders have characteristic features that are not shared with CDG, which can help identify the correct disorder. Screening tests for these diseases, along with CDG, are often performed early in assessment20.
Management
The broad clinical spectrum presented by individuals affected by CDG generally leads disease management to be determined on an individual’s specific needs, often requiring multidisciplinary care from occupational, speech, and physical therapists and neurological, endocrinological, hematological and gastroenterological specialists. CDG management can involve an array of strategies, as documented by Krasnewich et al. (2021), such as20:
- Coagulopathy prevention – which can involve patient and family education of risk and clinical presentation of thrombosis and consultation with hematologist when appropriate
- Failure to thrive management – which can involve use of special formulas to maximize caloric intake, as required, and management for individuals with oral motor defects, such as thickening of liquids
- Life skills and vocational training – as appropriate, to aid adult independence in appropriate individuals
- Rehabilitation therapy – which can involve a combination of occupational, speech, feeding and physical therapy
- Ophthalmological therapy – which may involve management provided by ophthalmologist
- Musculoskeletal management – which can involve corrective surgeries (in severe cases)
- Vaccination
In addition, individuals with CDG can require regular assessment to monitor organ and body system functioning, such as tests to monitor liver and thyroid function, urinalysis, serum gonadotropins, protein C, protein S, antithrombin III, factor IX, as well as echocardiogram, kidney ultrasound, ophthalmologic and clinical genetics assessments20.
Treatment
The most common therapeutic strategy for CDG are dietary supplements, although treatment of CDG predominantly involves managing symptoms21. These supplements are typically in the form of monosaccharides, including mannose, for MPI-CDG; fucose, for SLC35C1-CDG; sialic acid and N-acetlymannosamine, for GNE-CDG (clinical studies); and galactose, for PGM1-CDG (clinical trials) and TMEM165-CDG. Uridine supplements have been used for CAD-CDG treatment and butyrate supplements have been used for PIGM-CDG treatment (one individual)21. In addition to dietary supplements, some CDG have been shown to be treated with liver (MPI-CDG, CCDC115-CDG and ATP6AP1-CDG), heart (DOLK-CDG) or bone marrow transplants (PGM3-CDG), although success rates can vary21. In addition, a number of therapeutic strategies for CDG are being explored at various stages of development, such as repurposed drugs (such as acetazolamide and epalrestat), pharmacological chaperones, antisense therapy and gene therapy22.
Patient Registries
There are CDG patient registries across the globe that include patients of all CDG types.
CDG Connect Patient Insights Network (PIN)
Patients or their caregivers can enroll directly in the CDG Connect patient registry.
CDG Connect is a confidential patient registry that collects and organizes clinical information from CDG patients across the globe that patients of their caregivers can directly enroll in. The registry aims to inform and advance research and understanding of CDG, helping researchers develop new treatments and improving the lives of CDG patients. By enrolling, patients and caregivers can also learn about new and emerging CDG therapies, clinical trials and current progress in the field.
Unified European Registry for Inherited Metabolic Disorders (U-IMD)
The U-IMD patient registry is available for members of the European Reference Network for Hereditary Metabolic Disorders (MetabERN) and for collaborating health care providers outside of MetabERN.
U-IMD is a registry that collects information on patients affected with inherited metabolic disorders (such as CDG) and aims to promote the health and improve the lives of these patients. U-IMD is intended to be used by physicians treating patients with rare inherited metabolic disorders.
CDG CARE and the University of Utah have developed a patient registry to collect vital data from all patients diagnosed with CDG types that fall under the classification of GPI Anchor Deficiencies.
Resources
For Healthcare Professionals
IEMbase: Inborn Errors of Metabolism Knowledgebase
An online expert-curated knowledgebase of 530+ inborn errors of metabolism to support clinical communities in the diagnosis of IEMs.
Publications
- Krasnewich DM, Lam C, Ferreira C. Overview of Congenital Disorders of Glycosylation. UpToDate; Retrieved September 23 2021.
- Lipiński P, Tylki-Szymańska A. Congenital Disorders of Glycosylation: What Clinicians Need to Know? Frontiers in Pediatrics. 2021; 9. doi: 10.3389/fped.2021.715151
- Ferreira CR, Altassan R, Marques-Da-Silva D. et al. Recognizable phenotypes in CDG. Journal of Inherited Metabolic Disease. 2018; 41 (3). doi: 10.1007/s10545-018-0156-5
- Johnsen C, Edmondson AC. Manifestations and Management of Hepatic Dysfunction in Congenital Disorders of Glycosylation. Clinical Liver Disease. 2021; 18 (2). doi: 10.1002/cld.1105
- Park JH, Marquardt T. Treatment Options in Congenital Disorders of Glycosylation. Frontiers in Genetics. 2021. doi: 10.3389/fgene.2021.735348
- Boyer SW, Johnsen C, Morava E. Nutrition interventions in congenital disorders of glycosylation. Trends in Molecular Medicine. 2022; 28 (6). doi: 10.1016/j.molmed.2022.04.003
- Altassan R, Peanne R, Jaeken J, et al. International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: Diagnosis, treatment and follow-up. Journal of Inherited Metabolic Disease. 2019; 42 (1). doi: 10.1002/jimd.12024
- Altassan R, Radenkovic S, Edmondson AC, et al. International consensus guidelines for phosphoglucomutase 1 deficiency (PGM1-CDG): Diagnosis, follow-up, and management. Journal of Inherited Metabolic Disease. 2021; 44 (1). doi: 10.1002/jimd.12286
- Čechová A, Altassan R, Borge D, et al. Consensus guideline for the diagnosis and management of mannose phosphate isomerase-congenital disorder of glycosylation. Journal of Inherited Metabolic Disease. 2020; 43 (4). doi: 10.1002/jimd.12241
- Lam C, Wolfe L, Need A, et al. NGLY1-Related Congenital Disorder of Deglycosylation. GeneReviews® [Internet]. 2018
Journal of Inherited Metabolic Disease Podcasts
A fortnightly podcast from the Journal of Inherited Metabolic Disease, where authors discuss recent publications from the journal. The podcast is intended for specialists and interested clinicians but is also intended to increase the accessibility of this work for patients and families.
For Patients, Families and Caregivers
A number of educational resources for new patients can be found on the Resources Page.
The Patient Education Guide "Congenital Disorders of Glycosylation (CDG)" developed by the Mayo Clinic can be found here.
The Patient Education Guide “GPI Anchor Disorders: A Subtype of Congenital Disorders of Glycosylation” developed by the Mayo Clinic can be found here.
References
- Péanne R, de Lonlay P, Foulquier F, et al. Congenital disorders of glycosylation (CDG): Quo vadis? European Journal of Medical Genetics. 2018;61(11). doi:10.1016/j.ejmg.2017.10.012
- Ondruskova N, Cechova A, Hansikova H, Honzik T, Jaeken J. Congenital disorders of glycosylation: Still “hot” in 2020. Biochimica et Biophysica Acta (BBA) - General Subjects. 2021;1865(1). doi:10.1016/j.bbagen.2020.129751
- Reily C, Stewart TJ, Renfrow MB, Novak J. Glycosylation in health and disease. Nature Reviews Nephrology. 2019;15(6). doi:10.1038/s41581-019-0129-4
- Paprocka J, Jezela-Stanek A, Tylki-Szymańska A, Grunewald S. Congenital Disorders of Glycosylation from a Neurological Perspective. Brain Sciences. 2021;11(1). doi:10.3390/brainsci11010088
- Francisco R, Marques-da-Silva D, Brasil S, et al. The challenge of CDG diagnosis. Molecular Genetics and Metabolism. 2019;126(1). doi:10.1016/j.ymgme.2018.11.003
- Briones P, Vilaseca MA, Garcı́a-Silva MT, et al. Congenital disorders of glycosylation (CDG) may be underdiagnosed when mimicking mitochondrial disease. European Journal of Paediatric Neurology. 2001;5(3). doi:10.1053/ejpn.2001.0483
- Supraha Goreta S, Dabelic S, Dumic J. Insights into complexity of congenital disorders of glycosylation. Biochemia Medica. Published online 2012. doi:10.11613/BM.2012.019
- Lipiński P, Bogdańska A, Tylki-Szymańska A. Congenital disorders of glycosylation: Prevalence, incidence and mutational spectrum in the Polish population. Molecular Genetics and Metabolism Reports. 2021;27. doi:10.1016/j.ymgmr.2021.100726
- Alsubhi S, Alhashem A, Faqeih E, et al. Congenital disorders of glycosylation: The Saudi experience. American Journal of Medical Genetics, Part A. 2017;173(10). doi:10.1002/ajmg.a.38358
- Chang IJ, He M, Lam CT. Congenital disorders of glycosylation. Annals of Translational Medicine. 2018;6(24). doi:10.21037/atm.2018.10.45
- Jaeken J, Hennet T, Matthijs G, Freeze HH. CDG nomenclature: Time for a change! Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2009;1792(9). doi:10.1016/j.bbadis.2009.08.005
- Raynor A, Vincent-Delorme C, Alaix A-S, et al. Normal transferrin patterns in congenital disorders of glycosylation with Golgi homeostasis disruption: apolipoprotein C-III at the rescue! Clinica Chimica Acta. 2021;519. doi:10.1016/j.cca.2021.05.016
- Lefeber DJ. Protein-Specific Glycoprofiling for Patient Diagnostics. Clinical Chemistry. 2016;62(1). doi:10.1373/clinchem.2015.248518
- Lefeber DJ, Morava E, Jaeken J. How to find and diagnose a CDG due to defective N-glycosylation. Journal of Inherited Metabolic Disease. 2011;34(4). doi:10.1007/s10545-011-9370-0
- Asteggiano CG, Papazoglu M, Bistué Millón MB, et al. Ten years of screening for congenital disorders of glycosylation in Argentina: case studies and pitfalls. Pediatric Research. 2018;84(6). doi:10.1038/s41390-018-0206-6
- Magalhães APPS de, Burin MG, Souza CFM de, et al. Transferrin isoelectric focusing for the investigation of congenital disorders of glycosylation: analysis of a ten-year experience in a Brazilian center. Jornal de Pediatria. 2020;96(6). doi:10.1016/j.jped.2019.05.008
- Thiel C, Meßner-Schmitt D, Hoffmann GF, Körner C. Screening for congenital disorders of glycosylation in the first weeks of life. Journal of Inherited Metabolic Disease. 2013;36(5). doi:10.1007/s10545-012-9531-9
- Čechová A, Altassan R, Borgel D, et al. Consensus guideline for the diagnosis and management of mannose phosphate isomerase‐congenital disorder of glycosylation. Journal of Inherited Metabolic Disease. 2020;43(4). doi:10.1002/jimd.12241
- Jones MA, Rhodenizer D, da Silva C, et al. Molecular diagnostic testing for congenital disorders of glycosylation (CDG): Detection rate for single gene testing and next generation sequencing panel testing. Molecular Genetics and Metabolism. 2013;110(1-2). doi:10.1016/j.ymgme.2013.05.012
- Krasnewich DM, Lam C, Ferreira C. Overview of Congenital Disorders of Glycosylation. (Hahn S, TePas E, eds.). UpToDate; 2021.
- Sosicka P, Ng BG, Freeze HH. Congenital Disorders of Glycosylation. In: Comprehensive Glycoscience. Elsevier; 2021. doi:10.1016/B978-0-12-819475-1.00013-4
- Brasil S, Pascoal C, Francisco R, et al. CDG Therapies: From Bench to Bedside. International Journal of Molecular Sciences. 2018;19(5). doi:10.3390/ijms19051304
- Abu Bakar, N., Lefeber, D. J. & van Scherpenzeel, M. Clinical glycomics for the diagnosis of congenital disorders of glycosylation. J. Inherit. Metab. Dis. 41, (2018).