
Lay Summary
ALG13-CDG, formerly known as CDG-Is, is a rare genetic condition that affects many systems in the body. To date, there are 60 patients with ALG13-CDG reported in the literature. ALG13-CDG is classified as a disorder of N-linked protein glycosylation. ALG13-CDG is caused when a mutation arises spontaneously in an individual's ALG13 gene (located on the X chromosome), which provides instructions for making an enzyme that attaches the simple sugar N-acetylglucosamine to growing sugar chains during N-glycosylation. Mutations in the ALG13 gene cause proteins to have incomplete or absent sugar chains. The majority of patients with ALG13-CDG are female, although a few cases in males have been reported. Symptoms of ALG13-CDG begin at infancy and are primarily characterized by early onset epilepsy and global developmental delay in females and epilepsy, isolated developmental delay and liver abnormalities in males. Screening tests typically used for CDG-I type are not reliable in ALG13-CDG patients and diagnosis is achieved through genetic testing. There are currently no approved treatments for ALG13-CDG. Treatment is focused on management of specific symptoms and preventing complications.
Overview
UDP-N-acetylglucosamine (UDP-GlcNAc) transferase subunit congenital disorders of glycosylation (ALG13-CDG) is a rare X-linked genetic disorder. The first reported case of ALG13-CDG was in 20121 and to date, 60 cases have been reported1–15. The ALG13 (asparagine-linked glycosylation 13) gene encodes a UDP-N-acetylglucosamine transferase enzyme responsible for adding the second N-Acetylglucosamine (GlcNAc) residue during synthesis of the lipid-linked oligosaccharide (LLO) in the endoplasmic reticulum (ER). LLO is a precursor step to N-glycosylation, which is the process by which sugar chains (glycans) are added to the amino acid asparagine in some proteins. Deficiency in the ALG13 enzyme results in the incomplete assembly of the LLO, leading to insufficient N-glycosylation of proteins.
Symptoms begin at infancy and can differ between males and females. The characteristic presentation of ALG13-CDG in females include early onset epileptic encephalopathy (within the first 6 months), and mild to severe global developmental delay14,16. In males the characteristic presentation include mild seizures and isolated developmental delay, liver abnormalities, recurrent infections, and ophthalmological problems17. Males’ biochemical analyses show abnormal coagulation factors and transferrin glycosylation, while females’ biochemical analyses show no abnormalities17. A diagnosis can only be determined through molecular genetic testing, as screening tests typically used for CDG-I patients are normal for most ALG13-CDG patients14,15,18. There are currently no approved treatments for ALG13-CDG.
Synonyms
- CDG-Is
- CDG-1S
- CDG syndrome type 1s
- Congenital disorder of glycosylation type 1s
- Epileptic encephalopathy, early infantile, 36
Inheritance
ALG13-CDG is an X-linked dominant disorder, meaning females can inherit one defective copy of the gene from one symptomatic parent (mother or father) and males can inherit one defective copy from their symptomatic mother. However, ALG13-CDG is usually caused by a new genetic mutation that arises in the affected individual (de novo) and is not inherited from a parent17.
Gene Function
The ALG13 gene encodes a subunit of a bipartite UDP-N-acetylglucosamine (GlcNAc) transferase. The ALG13 protein heterodimerizes with the protein encoded by ALG14 to form a functional UDP-GlcNAc glycosyltransferase. Glycosyltransferases are enzymes that transfer glycosyl molecules during glycosylation. ALG13 is in the ER membrane where it has a role in the assembly of the LLO, a precursor for protein N-glycosylation19.
The complete UDP-GlcNAc glycosyltransferase enzyme, containing the ALG13 and ALG14 protein subunits, transfers the second GlcNAc residue to the growing oligosaccharide prior to its attachment to a protein19.
LLO synthesis
N-glycosylation is the process by which an oligosaccharide is attached the nitrogen atom of asparagine residues on proteins. N-glycosylation is initiated in the ER and begins with the synthesis of the LLO 20. The LLO is comprised of a 14-sugar oligosaccharide, which is sometimes referred to as the N-glycan precursor, attached to the lipid carrier dolichol pyrophosphate (Dol-PP). The 14-sugar residues are two N-acetylglucosamine, nine mannose, and three glucose residues. Once assembled, the oligosaccharide is transferred “en bloc” to proteins and undergoes further processing in the ER and Golgi20. Once the oligosaccharide is attached to a protein, it is referred to as an N-glycan.
LLO synthesis is carried out by a series of enzymes encoded mostly by the ALG genes and can be divided into two phases: Phase I and Phase II (Figure 1)20.
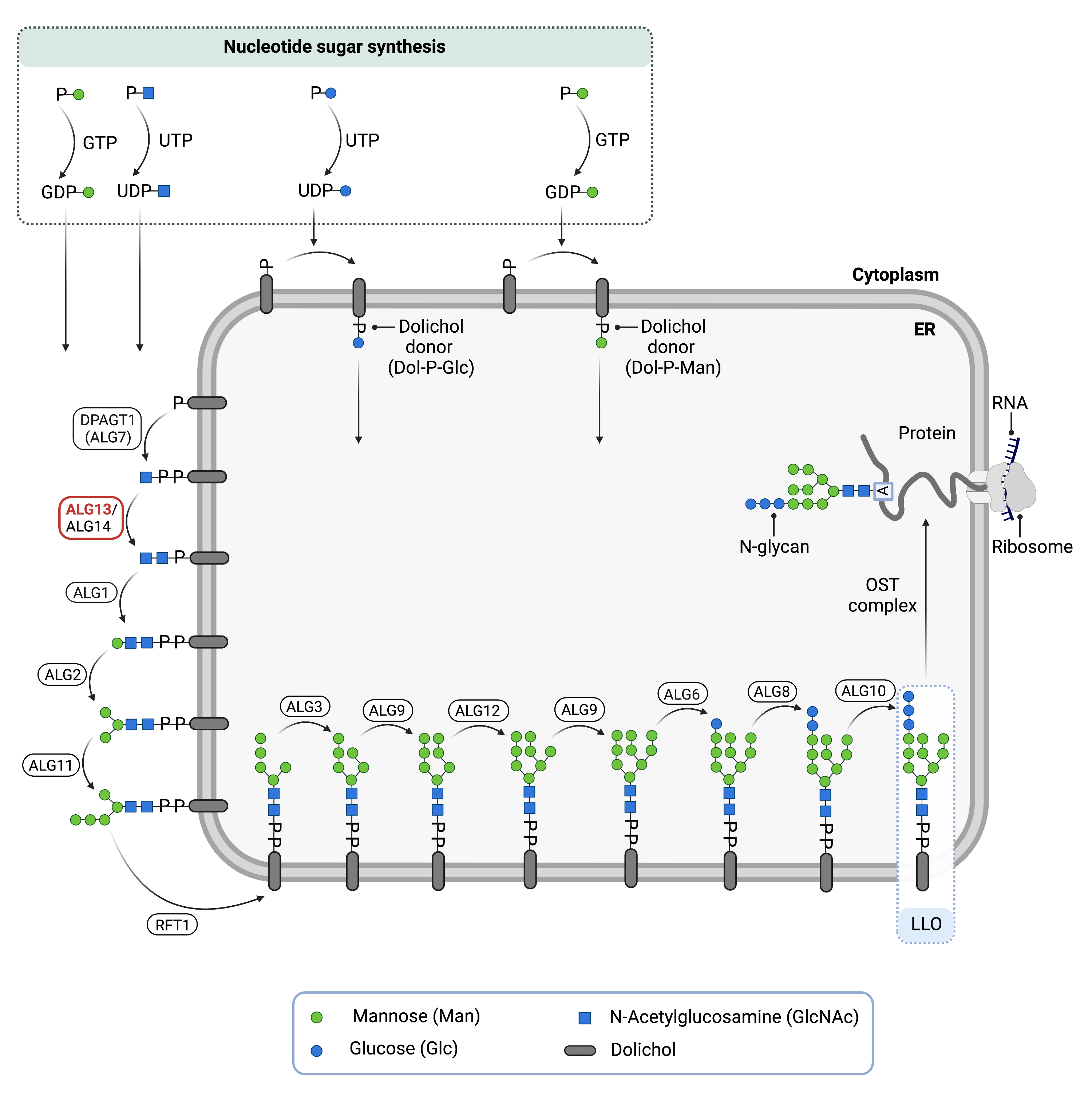
Figure 1. Role of ALG13 in glycosylation.
ALG13 is a subunit of an enzyme (UDP-N-acetylglucosamine (UDP-GlcNAc) transferase) involved in synthesising the lipid-linked oligosaccharide (LLO), which is needed for N-glycosylation. UDP-GlcNAc transferase adds a GlcNAc residue to the LLO at the endoplasmic reticulum surface.
Phase I
Phase I of LLO synthesis takes place on the cytoplasmic side of the ER. It begins with the sequential attachment of two N-Acetylglucosamine (GlcNAc2) and five mannose (Man5) residues to Dol-PP by several ER enzymes. GlcNAc and mannose are transferred from nucleotide sugars UDP-GlcNAc and GDP-Man, respectively. The intermediate structure, Dol-PP-GlcNAc2Man5, is translocated across the ER membrane into the lumen via the RFT1 enzyme20,21. ALG13 functions in this phase, adding the second GlcNAc residue to the growing LLO.
Phase II
Phase II of LLO synthesis takes place in the ER lumen. Once in the lumen, four Man residues followed by three glucose (Glc) residues are added to the intermediate structure, generating the complete the N-glycan, Dol-PP-Glc3Man9GlcNAc2. Man and Glc are transferred from glycosyl donors, Dol-P-Man and Dol-P-Glc, which are formed on the cytoplasmic side of the ER and must also be flipped across the ER membrane20.
Once assembled, the oligosaccharide is transferred “en bloc” from Dol-PP to asparagine residues of newly synthesized protein via the enzyme oligosaccharyltransferase (OST), resulting in N-glycosylation of the protein20. The asparagine residue must be located in regions of the protein where the sequence of amino acids is asparagine-X-serine/threonine, also called Asn-X-Ser/Thr consensus sequence, where “X” can be any amino acid except proline. The activity of OST is highly specific for the completely assembled 14-sugar N-glycan, Glc3Man9GlcNAc2.
Disease Mechanism
Mutations in the ALG13 gene lead to the production of an abnormal enzyme with reduced or no activity. In the absence of ALG13, it cannot form a functional UDP-GlcNAc glycosyltransferase enzyme with ALG14. This leads to the second GlcNAc not being added to the LLO, meaning the LLO assembly is incomplete. This results in reduced transfer efficiency of the oligosaccharide by OST, resulting in N-glycoproteins that are insufficiently glycosylated (hypoglycosylation).
Mutations in ALG13 may also contribute to the epileptic phenotype through interactions with GABAARa2 subunit of GABAA membrane receptors. GABA is a neurotransmitter in the central nervous system and studies in mice show that a deficiency in ALG13 affects transcription levels of GABAARa2, which may contribute to the epileptic phenotype observed in ALG13-CDG patients22.
Mutations
The ALG13 gene is found on the X chromosome (Xq23). The majority of individuals affected by ALG13-CDG have the mutation variant p.(Asn107Ser), with few other de novo mutations being reported14. The most common heterozygote mutation in females is the c.320A>G (p.Asn107Ser) variant23.
Signs & Symptoms
Clinical Presentation
Individuals with ALG13-CDG typically develop signs and symptoms during infancy. ALG13-CDG is primarily characterized by developmental and epileptic encephalopathy. ALG13-CDG is an X-linked condition and can affect males and females differently.
The characteristic clinical presentations of ALG13-CDG in females include2,14,16,17:
- Neurological – epileptic spasms with onset within the first 6 months of life (epileptic encephalopathy, early infantile, 36 (EEI36)), hypsarrhythmia in infancy, tonic seizures, global developmental delay, speech delay, motor delay, impaired eye contact, low muscle tone (hypotonia), and smaller than average head size (microcephaly)
90% of patients also have West syndrome. Dysmorphic features are a less common clinical presentation of ALG13-CDG, including micrognathia and hypertelorism14.
The characteristic clinical presentations of ALG13-CDG in males include15,17,24:
- Neurological – seizures, isolated developmental delay (delayed motor and speech development), and intellectual disability
- Liver involvement – enlarged liver (hepatomegaly)
- Immunological – recurrent infections
- Ophthalmological – nystagmus and bilateral optic nerve atrophy
Males have also been reported with normal cognitive development24.
Biochemical Abnormalities
In males, elevated liver enzymes, abnormal coagulation factors, and abnormal transferrin glycosylation have been reported17; however, some males have also been reported with normal transferrin glycosylation24. Females show normal transferrin glycosylation and do not typically have abnormal coagulation profiles17.
Classification
ALG13-CDG is classified as a disorder of N-linked protein glycosylation.
Under the former CDG classification system, ALG13-CDG is classified as a Type I CDG, which arise due to defects in the synthesis of oligosaccharides or their transfer to proteins.
Diagnosis
Although diagnosis of ALG13-CDG may be suspected based on presentation of symptoms and a detailed patient history, direct molecular genetic testing is the only definitive diagnostic test. Screening in suspected patients begins with a blood test to analyze serum transferrin. However, some ALG13-CDG patients can have normal results following transferrin isoelectric focusing (TIEF), and so transferrin analysis it is not a reliable diagnostic technique for ALG13-CDG. Direct molecular genetic testing is required for diagnosis. Potential biomarkers have been identified but require further investigation.
Transferrin Analysis
Individuals with ALG13-CDG show normal or mildly abnormal patterns by TIEF and thus TIEF is not reliable for diagnosis and other biomarkers are needed14,15,18.
Biomarkers
Three metabolites have been identified as potential biomarkers to aid in diagnosis of ALG13-CDG: betaine, N-acetyl-glycoprotein, and carnitine18.
Prognosis
Prognosis of ALG13-CDG patients is not well understood and may vary depending on the severity of an individual’s symptoms. Severely affected patients may have drug-resistant seizures17.
Management
Management of symptoms may include combinations of physical therapy, occupational therapy, speech or vision therapy and palliative measures. Epilepsy has been reported to be drug resistant but has been managed through ketogenic diet, oral steroids, vigabatrin, or adrenocorticotropic hormone14,16,17.
Therapies
There are currently no treatment options available for ALG13-CDG. Treatment is focused on management of symptoms and prevention of complications.
Research Models
Several ALG13 research models have been generated including yeast and mouse model organisms.
Yeast (S. cerevisiae)
Alg13 mutants have reduced function and show slow growth, defective protein glycosylation and accumulate LLO’s with one GlcNAc residue (YeastGenome)25.
Mouse (M. musculus)
Alg13 knockout mouse
Homozygous whole body Alg13 knockout mice have been generated from the mouse line Alg13tm1a(KOMP)Wtsi. Mutants live to early adulthood and phenotypically show decreased lean body mass, increased total body fat amount, increased leukocyte cell number, decreased bone mineral content and decreased bone mineral density (IMPC).
ALG13 knockout mice have been used to characterize the epileptic phenotype of ALG13-CDG patients. ALG13KO mice display spontaneous seizures and epileptic activity, and a decrease in cortical inhibitory synaptic transmission. It was found that ALG13 influences epileptic activity through interactions with GABA receptor subunits22.
Alg6 conditional-ready floxed knockout embryonic stem cell line
Embryonic Stem (ES) cell line Alg13tm1a(KOMP)Wtsi is a targeted knockout/null mutation of Alg13. A “conditional-ready” allele can be created by flp recombinase expression in mice carrying this allele, and cre expression results in a knockout mouse. If cre is expressed without flp expression, a reporter knockout mouse can be generated (MGI).
ALG6 knockout ES cell line
ES cell line Alg13tm1a(EUCOMM)Hmgu and Alg13tm1e(KOMP)Wtsi are targeted knockout/null mutations of Alg13 (MGI, MGI).
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including ALG13-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
GROUPS
Publications
ALG13-CDG Scientific Articles on PubMed
Additional Resources
ALG13-CDG on FCDGC
OMIM
Orphanet
GARD
NORD
Genetic Testing Registry
ClinVar
NIH
GeneCards
UniProt
Epilepsy Genetics
References
- Timal, S. et al. Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Human Molecular Genetics 21, (2012).
- Madaan, P., Negi, S., Sharma, R., Kaur, A. & Sahu, J. K. X-Linked ALG13 Gene Variant as a Cause of Epileptic Encephalopathy in Girls. The Indian Journal of Pediatrics 86, (2019).
- Yuskaitis, C. J. et al. Infantile Spasms of Unknown Cause: Predictors of Outcome and Genotype-Phenotype Correlation. Pediatric Neurology 87, (2018).
- Bastaki, F. et al. Single‐center experience of N‐linked Congenital Disorders of Glycosylation with a Summary of Molecularly Characterized Cases in Arabs. Annals of Human Genetics 82, (2018).
- Fung, C.-W., Kwong, A. K.-Y. & Wong, V. C.-N. Gene panel analysis for nonsyndromic cryptogenic neonatal/infantile epileptic encephalopathy. Epilepsia Open 2, (2017).
- Zhu, X. et al. A case-control collapsing analysis identifies epilepsy genes implicated in trio sequencing studies focused on de novo mutations. PLOS Genetics 13, (2017).
- Ortega-Moreno, L. et al. Molecular diagnosis of patients with epilepsy and developmental delay using a customized panel of epilepsy genes. PLOS ONE 12, (2017).
- Geisheker, M. R. et al. Hotspots of missense mutation identify neurodevelopmental disorder genes and functional domains. Nature Neuroscience 20, (2017).
- Hamici, S., Bastaki, F. & Khalifa, M. Exome sequence identified a c.320A > G ALG13 variant in a female with infantile epileptic encephalopathy with normal glycosylation and random X inactivation: Review of the literature. European Journal of Medical Genetics 60, (2017).
- Myers, C. T. et al. De Novo Mutations in SLC1A2 and CACNA1A Are Important Causes of Epileptic Encephalopathies. The American Journal of Human Genetics 99, (2016).
- Kobayashi, Y. et al. High prevalence of genetic alterations in early-onset epileptic encephalopathies associated with infantile movement disorders. Brain and Development 38, (2016).
- Dimassi, S. et al. Whole‐exome sequencing improves the diagnosis yield in sporadic infantile spasm syndrome. Clinical Genetics 89, (2016).
- Michaud, J. L. et al. The genetic landscape of infantile spasms. Human Molecular Genetics 23, (2014).
- Datta, A. N. et al. The phenotypic spectrum of X‐linked, infantile onset ALG13 ‐related developmental and epileptic encephalopathy. Epilepsia 62, (2021).
- Galama, W. H., Verhaagen – van den Akker, S. L. J., Lefeber, D. J., Feenstra, I. & Verrips, A. ALG13-CDG with Infantile Spasms in a Male Patient Due to a De Novo ALG13 Gene Mutation. in (2017). doi:10.1007/8904_2017_53.
- Ng, B. G. et al. Predominant and novel de novo variants in 29 individuals with ALG13 deficiency: Clinical description, biomarker status, biochemical analysis, and treatment suggestions. J Inherit Metab Dis 43, 1333–1348 (2020).
- ALG13 Congenital Disorder of Glycosylation | Rare Diseases Clinical Research Network. https://www.rarediseasesnetwork.org/fcdgc/alg13.
- Paprocka, J. et al. The First Metabolome Analysis in Children with Epilepsy and ALG13-CDG Resulting from c.320A>G Variant. Children 8, (2021).
- Gao, X.-D., Tachikawa, H., Sato, T., Jigami, Y. & Dean, N. Alg14 Recruits Alg13 to the Cytoplasmic Face of the Endoplasmic Reticulum to Form a Novel Bipartite UDP-N-acetylglucosamine Transferase Required for the Second Step of N-Linked Glycosylation. Journal of Biological Chemistry 280, (2005).
- Bieberich, E. Synthesis, Processing, and Function of N-glycans in N-glycoproteins. in (2014). doi:10.1007/978-1-4939-1154-7_3.
- Stanley, P., Taniguchi, N. & Aebi, M. N-glycans. in Essentials of Glycobiology [Internet] (eds. Varki, A. et al.) (Cold Spring Harbor Laboratory Press, 2017).
- Huo, J. et al. ALG13 participates in epileptogenesis via regulation of GABAA receptors in mouse models. Cell Death Discovery 6, (2020).
- Mitusińska, K. et al. Structural Analysis of the Effect of Asn107Ser Mutation on Alg13 Activity and Alg13-Alg14 Complex Formation and Expanding the Phenotypic Variability of ALG13-CDG. Biomolecules 12, 398 (2022).
- Gadomski, T. E. et al. ALG13-CDG in a male with seizures, normal cognitive development, and normal transferrin isoelectric focusing. Am J Med Genet A 173, 2772–2775 (2017).
- Bickel, T., Lehle, L., Schwarz, M., Aebi, M. & Jakob, C. A. Biosynthesis of Lipid-linked Oligosaccharides in Saccharomyces cerevisiae. Journal of Biological Chemistry 280, 34500–34506 (2005).