
Lay Summary
RFT1-CDG, formerly known as CDG-In, is a rare inherited condition that affects many parts of the body. To date, 16 cases of RFT1-CDG have been reported in the literature. RFT1-CDG is classified as a disorder of N-linked protein glycosylation. RFT1-CDG is caused when an individual has mutations in both copies of the RFT1 gene, which provides instructions for making an enzyme that is involved in N-glycosylation of proteins. The RFT1 enzyme flips a lipid-sugar molecule (Dol-PP-Man5GlcNAc2) from the cytosol to the inside of the endoplasmic reticulum where additional sugars can be added. The completed sugar chain (glycan) is then transferred to proteins during N-glycosylation. Mutations in the RFT1 gene cause proteins to be under glycosylated, meaning proteins have abnormal or lower levels of sugars attached. Symptoms of RFT1-CDG begin at infancy and are primarily characterized by neurological abnormalities, including developmental delay, seizures, and deafness that typically presents in newborns to young children. Several screening tests are available for RFT1-CDG, but a definitive diagnosis is achieved through genetic testing. Treatment is focused on the management of specific symptoms and preventing complications.
Overview
RFT1 congenital disorder of glycosylation (RFT1-CDG) is a rare autosomal recessive genetic disorder. The first reported case of RFT1-CDG was in 20001 , and there is a total of 16 cases reported in the literature so far1–14. The RFT1 gene encodes an enzyme, RTF1, which is needed for lipid-linked oligosaccharide (LLO) synthesis during N-glycosylation of proteins. RTF1 is responsible for transferring a LLO precursor, Dol-PP-Man5GlcNAc2, from the cytosolic side of the endoplasmic reticulum (ER) to the lumen side of the ER during LLO synthesis. Deficiency in the RFT1 enzyme results in an accumulation of the Dol-PP-Man5GlcNAc2 precursor and insufficient N-glycosylation of proteins. Symptoms begin at infancy and the characteristic presentations of RFT1-CDG include developmental delay, seizures, and deafness. A diagnosis can be determined through transferrin analysis and LLO analysis. However, a definitive diagnosis can only be achieved through molecular genetic testing. There are currently no approved treatments for RFT1-CDG.
Synonyms
- CDG syndrome type In
- Congenital disorder of glycosylation type In
- CDG-In
- CDGIN
- Lipid-linked oligosaccharide flippase deficiency
- Carbohydrate deficient glycoprotein syndrome type In
- Man5GlcNAc2-PP-Dol flippase deficiency
- Congenital disorder of glycosylation type 1n
Inheritance
RFT1-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene function
RFT1 encodes a putative flippase enzyme, RFT1. RFT1 is responsible for transferring Dol-PP-Man5GlcNAc2 from the cytoplasmic side to the luminal side of the ER during LLO synthesis11,13–15.
LLO Synthesis
N-glycosylation is the process by which an oligosaccharide is attached to a nitrogen atom of an asparagine residue on a protein. N-glycosylation is initiated in the ER and begins with the synthesis of the LLO16. The LLO is comprised of a 14-sugar oligosaccharide attached to the lipid carrier: dolichol pyrophosphate (Dol-PP). The 14-sugar glycan chain is made up of two N-acetylglucosamine (GlcNAc), nine mannose, and three glucose residues. Once assembled, the oligosaccharide is transferred “en bloc” to a protein and undergoes further processing in the ER and Golgi. Once the oligosaccharide is attached to the asparagine residue of a protein, it is referred to as an N-glycan16.
LLO synthesis is carried out by a series of enzymes encoded by the DPAGT1 and ALG genes and can be divided into two phases: Phase I and Phase II (Figure 1)16.
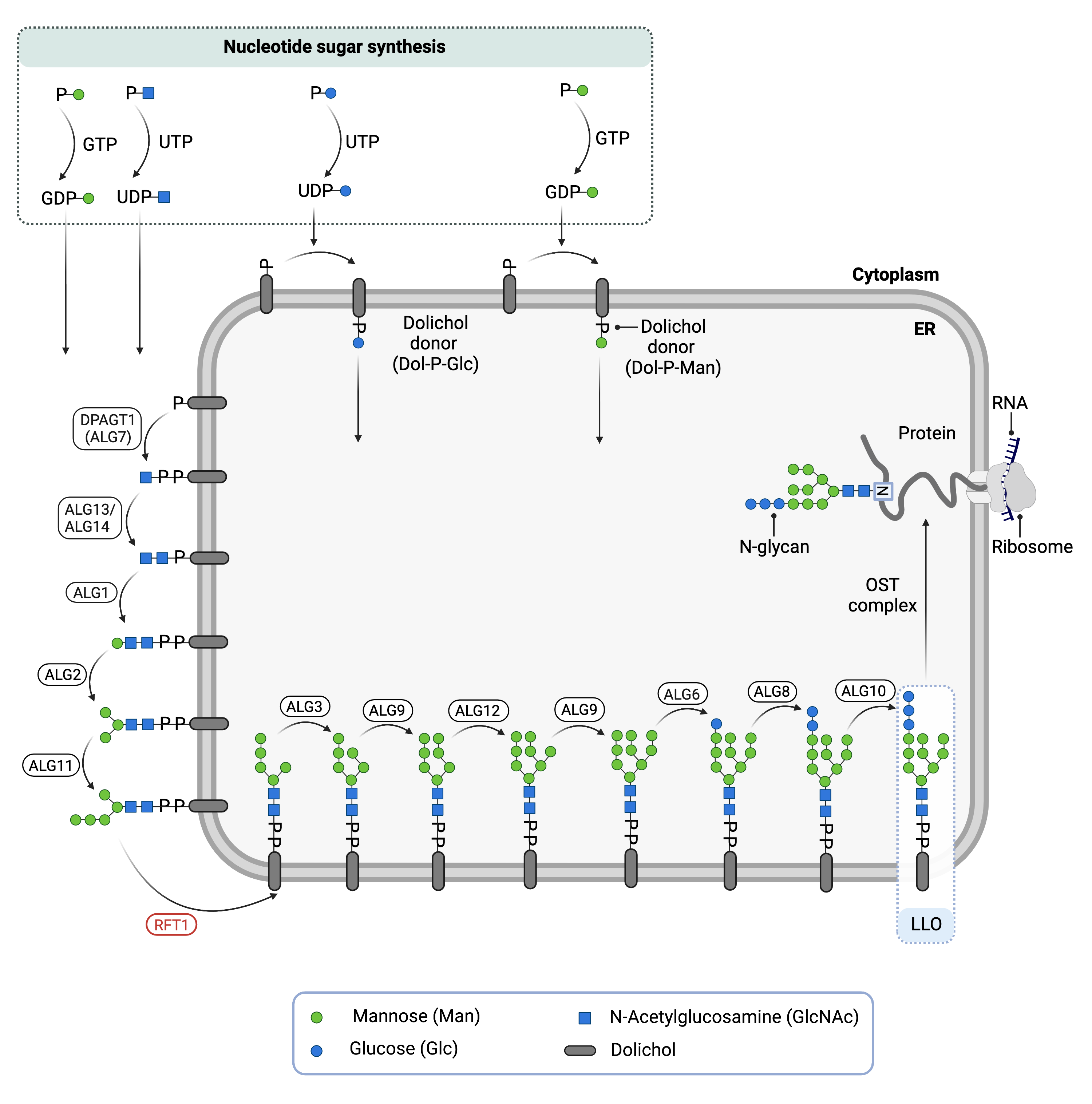
Figure 1. Role of RFT1 in glycosylation.
RFT1 is an enzyme involved in lipid-linked oligosaccharide (LLO) synthesis, which is needed for N-glycosylation. RFT1 flips the LLO precursor, Dol-PP-Man5GlcNAc2, from the cytosolic side of the ER to the lumen side of the ER.
Phase I
Phase I of LLO synthesis takes place on the cytoplasmic side of the ER. It begins with the sequential attachment of two N-acetylglucosamine (GlcNAc2) and five mannose (Man5) residues to Dol-PP by several ER enzymes. GlcNAc and mannose are transferred from nucleotide sugars UDP-acetylglucosamine and GDP-mannose, respectively. The intermediate structure, Dol-PP-GlcNAc2Man5, is translocated from the cytoplasm into the ER lumen by the RTF1 enzyme (Figure 1)16,17.
Phase II
Phase II of LLO synthesis takes places in the ER lumen. Once in the lumen, four mannose residues followed by three glucose (Glc3) residues are added to the intermediate structure, generating the complete LLO structure: Dol-PP-GlcNAc2Man9Glc3. Mannose and glucose are transferred from glycosyl donors Dol-P-mannose and Dol-P-glucose, which are formed on the cytoplasmic side of the ER and must also be flipped across the ER membrane16.
Once assembled, the oligosaccharide is transferred “en bloc” from Dol-PP to an asparagine residue of the newly synthesized protein via the enzyme oligosaccharyltransferase (OST), resulting in N-glycosylation of the protein16. The activity of OST is highly specific for the completely assembled 14-sugar oligosaccharide, Glc3Man9GlcNAc2.
Disease Mechanism
Mutations in RFT1 lead to the RFT1 enzyme to lack or completely lose activity, which prevents the transfer of the Dol-PP-Man5GlcNAc2 precursor from the cytosolic side of the ER to the lumen side of the ER. Consequently, deficient RTF1 can cause an accumulation of the LLO precursor, Dol-PP-GlcNAc2Man5, on the cytosolic side of the ER and hypoglycosylation of proteins during N-glycosylation9,13,15,18.
Mutations
The RFT1 gene is located on Chromosome 3 (3q21.1). Missense mutations and point mutations have been reported. Two missense mutations, c.73C>T and c.208T>C have been reported to have a less severe phenotype compared to other patients in the literature11.
Signs & Symptoms
Clinical Presentation
Individuals with RFT1-CDG typically develop signs and symptoms during infancy. RFT1-CDG is primarily characterized by developmental delay, low muscle tone, and seizures. The characteristic clinical presentations of RFT1-CDG include2,6–12,14,19:
- Cardiovascular symptoms – thrombotic complications and excessive bleeding or clotting (coagulopathy)
- Developmental symptoms – global developmental delay, seizures, intellectual disability, respiratory insufficiency, sensorineural deafness, decreased vision, and dysmorphic features that can affect different parts of the body, such as the spine (kyphoscoliosis), mouth, nose, ears, eyes, hands, face, neck, hands, chest, and feet
- Growth symptoms – reduced muscle mass (hypotonia), failure to thrive, feeding problems, and enlarged liver
One case has also been reported with symptoms developing during pregnancy such as floppy neonate, fetal growth restriction, facial dysmorphism, severe respiratory insufficiency and ventilator dependence as a newborn. Multiple cases also had coagulation disorders, such as thrombosis and thrombophilia12.
Biochemical Abnormalities
Patients typically present a type I serum transferrin pattern and elevated Dol-PP-Man5GlcNAc2 levels, and can have decreased blood clotting factor XI, antithrombin, and protein C levels8,11.
Classification
RFT1-CDG is classified as a disorder of N-linked protein glycosylation.
Under the former CDG classification system, RFT1-CDG is classified as a Type I CDG, which arises due to defects in the synthesis of oligosaccharides or their transfer to N-glycoproteins.
Diagnosis
Although diagnosis of RFT1-CDG may be suspected based on presentation of symptoms and a detailed patient history, direct molecular genetic testing is the only definitive diagnostic test. First-line screening tests in suspected patients are serum transferrin analysis and LLO analysis. However, genetic testing via next-generation sequencing is required for a definitive diagnosis of RTF1-CDG.
Transferrin Analysis
Individuals with RFT1-CDG show a type I pattern by transferrin isoelectric focusing (TIEF). Type 1 patterns are observed in CDG that arise due to defects in the LLO synthesis and are characterized by an increase in disialo- and asialo-transferrin isoforms14.
LLO Analysis
LLO analysis of RFT1-CDG patient fibroblasts show an accumulation of the intermediate structure Dol-PP-Man5GlcNAc2, along with normal protein-linked glycans, which is expected for a defect in the translocation of Dol-PP-Man5GlcNAc2 to the ER lumen14.
Biomarkers
Accumulation of the intermediate LLO molecule, Dol-PP-GlcNAc2Man5 in patient fibroblasts may be used as a biomarker for RFT1-CDG11.
Prognosis
Prognosis of RFT1-CDG may vary depending on the severity of an individual’s symptoms and the type of mutation they harbor. As of 2019, a patient who is 18 years old has been reported in the medical literature11,12.
Management
Management of RFT1-CDG focuses on symptoms and avoiding complications. Management of symptoms may include combinations of physical and occupational therapy.
Therapies
Research Models
Several research models have been generated to RFT1-CDG, including yeast, protozoa and mouse models and human cells.
Yeast (S. cerevisiae)
In yeast, the Rft1 protein has been shown to mediate the translocation of the LLO across the ER membrane in a bidirectional manner15. However, there is evidence that suggest RFT1 has an accessory role in translocation, rather than acting as the Dol-PP-GlcNAc2Man5 flippase in yeast20.
Rft1-deficient yeast have been used to investigate the pathology of the p.R67C mutation, where it was found that the Rft1 deficiency leads to an accumulation of Dol-PP-GlcNAc2Man5 and hypoglycosylation of proteins13.
Yeast (H. polymorpha)
HpRFT1 was shown to have a role in translocating Dol-PP-GlcNAc2Man5 from the cytoplasmic side of the ER to the ER lumen in H. polymorpha. Overexpressing HpRFT1 increased growth in mutant strains Hpalg11Δ and Hpalg3Δalg11Δ, whereas strains lacking HpALG11 had poor growth21.
Protozoa (T. brucei)
TbRft1-null T. brucei cells grew well. TbRFT1-null cells had higher steady-state levels of mature LLOs than wildtype cells, and TbRft1-null cells also generated N-glycosylated proteins. Potentially, TbRft1 may act as a chaperone for Dol-PP-GlcNAc2 Man5in T. brucei, rather than acting as a flippase22.
In addition to N-glycosylation, TbRft1 was also found to play a role in glycosylphosphatidylinositol (GPI) anchor glycosylation in T. brucei. TbRft1 was also found to be localised to the Golgi, as well as the ER membrane23.
Mouse (M. musculus)
Rft1 reporter tagged deletion
Rft1tm1b(KOMP)Wtsi is a reporter tagged deletion mutation of Rft1 in mice. Mice generated with this knockout live from E9.5 to early adulthood, and phenotypically the mutation significantly affects mortality and the immune system (IMPC).
Human Cell Lines
Patient-derived fibroblasts
RFT1-CDG patient-derived fibroblasts produce monophosphorylated oligosaccharides (POS), which healthy control cells do not generate. Potentially, POS in RFT1-CDG arise from Dol-PP-Man5GlcNAc that has accumulated on the cytosolic side of the ER18.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including RFT1-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Publications
RFT1-CDG Scientific Articles on PubMed
Additional Resources
OMIM
IEMbase
Orphanet
GARD
NORD
Genetic Testing Registry
ClinVar
GeneCards
UniProt
References
- Imtiaz, F. et al. Genotypes and phenotypes of patients in the UK with carbohydrate-deficient glycoprotein syndrome type 1. Journal of Inherited Metabolic Disease 23, 162–174 (2000).
- Ondruskova, N. et al. RFT1-CDG in adult siblings with novel mutations. Mol Genet Metab 107, 760–762 (2012).
- Pérez-Cerdá, C. et al. A Population-Based Study on Congenital Disorders of Protein N- and Combined with O-Glycosylation Experience in Clinical and Genetic Diagnosis. The Journal of Pediatrics 183, 170-177.e1 (2017).
- Monies, D. et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Human Genetics 136, 921–939 (2017).
- Bastaki, F. et al. Single‐center experience of N‐linked Congenital Disorders of Glycosylation with a Summary of Molecularly Characterized Cases in Arabs. Annals of Human Genetics 82, 35–47 (2018).
- Barba, C. et al. Congenital disorders of glycosylation presenting as epileptic encephalopathy with migrating partial seizures in infancy. Developmental Medicine & Child Neurology 58, 1085–1091 (2016).
- Aeby, A. et al. RFT1-congenital disorder of glycosylation (CDG) syndrome: a cause of early-onset severe epilepsy. Epileptic Disorders 18, 92–96 (2016).
- Jaeken, J. et al. RFT1-CDG: Deafness as a novel feature of congenital disorders of glycosylation. Journal of Inherited Metabolic Disease 32, 335–338 (2009).
- Haeuptle, M. A. & Hennet, T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Human Mutation 30, 1628–1641 (2009).
- Clayton, P. T. & Grunewald, S. Comprehensive description of the phenotype of the first case of congenital disorder of glycosylation due to RFT1 deficiency (CDG In). Journal of Inherited Metabolic Disease 32, 137–139 (2009).
- Quelhas, D. et al. RFT1-CDG: Absence of Epilepsy and Deafness in Two Patients with Novel Pathogenic Variants. JIMD Rep 43, 111–116 (2019),
- Abiramalatha, T., Arunachal, G., Muthusamy, K. & Thomas, N. A family with floppy neonates with severe respiratory insufficiency: A lethal phenotype of RFT1-CDG due to a novel mutation. Eur J Med Genet 62, 248–253 (2019).
- Haeuptle, M. A. et al. Human RFT1 deficiency leads to a disorder of N-linked glycosylation. Am J Hum Genet 82, 600–606 (2008).
- Vleugels, W. et al. RFT1 Deficiency in Three Novel CDG Patients. Hum Mutat 30, 1428-1434 (2009).
- Helenius, J. et al. Translocation of lipid-linked oligosaccharides across the ER membrane requires Rft1 protein. Nature 2002 415:6870 415, 447–450 (2002).
- Bieberich, E. Synthesis, Processing, and Function of N-glycans in N-glycoproteins. in (2014).
- Stanley, P., Taniguchi, N. & Aebi, M. N-glycans. in Essentials of Glycobiology [Internet] (eds. Varki, A. et al.) (Cold Spring Harbor Laboratory Press, 2017).
- Vleugels, W. et al. Identification of phosphorylated oligosaccharides in cells of patients with a congenital disorders of glycosylation (CDG-I). Biochimie 93, (2011).
- OMIM Entry - # 612015 - CONGENITAL DISORDER OF GLYCOSYLATION, TYPE In; CDG1N.
- Rush, J. S., Cho, S. K., Jiang, S., Hofmann, S. L. & Waechter, C. J. Identification and characterization of a cDNA encoding a dolichyl pyrophosphate phosphatase located in the endoplasmic reticulum of mammalian cells. Journal of Biological Chemistry 277, (2002).
- Song, H., Qian, W., Wang, H. & Qiu, B. Identification and functional characterization of the HpALG11 and the HpRFT1 genes involved in N-linked glycosylation in the methylotrophic yeast Hansenula polymorpha. Glycobiology 20, (2010).
- Jelk, J. et al. Glycoprotein biosynthesis in a eukaryote lacking the membrane protein Rft1. Journal of Biological Chemistry 288, (2013).
- Gottier, P. et al. RFT1 protein affects glycosylphosphatidylinositol (GPI) anchor glycosylation. Journal of Biological Chemistry 292, (2017).