
Lay Summary
ALG8-CDG, formerly known as CDG-Ih, is a rare inherited condition that affects many parts of the body. There are 15 patients reported in the medical literature to date. ALG8-CDG is classified as a disorder of N-linked protein glycosylation. ALG8-CDG is caused by mutations in both copies of the ALG8 gene which encodes an enzyme that attaches the sugar glucose to sugar chains during N-glycosylation. Mutations in the ALG8 gene cause proteins to have abnormal sugars attached. Symptoms of ALG8-CDG begin at infancy and are primarily characterized by neurological abnormalities, including psychomotor delay and epilepsy, and problems with coordination and balance. Feeding difficulties, abnormal features, and gastrointestinal problems are also common. Several screening tests are available for ALG8-CDG, but a definitive diagnosis is achieved through genetic testing. There are currently no approved treatments for ALG8-CDG. Treatment is focused on the management of specific symptoms and preventing complications.
Overview
Alpha-1,3-glucosyltransferase congenital disorder of glycosylation (ALG8-CDG) is a rare autosomal recessive genetic disorder. The first reported case of ALG8-CDG was in 20032 and there are 15 reported cases to date1–8. The ALG8 (asparagine-linked glycosylation 8) gene encodes an enzyme responsible for adding the second glucose residue during synthesis of the lipid-linked oligosaccharide (LLO) in the endoplasmic reticulum (ER). LLO synthesis is a precursor step to N-glycosylation. Deficiency of the ALG8 enzyme results in the incomplete assembly of the LLO, leading to insufficient N-glycosylation of glycoproteins1.
Symptoms can first be detected prenatally, and the characteristic post-natal presentation of ALG8-CDG are neurological abnormalities, gastrointestinal and feeding problems, and dysmorphic features. A diagnosis can be determined through transferring analysis and LLO analysis, but a definitive diagnosis can only be achieved through molecular genetic testing1. There are currently no approved treatments for ALG8-CDG1.
Synonyms
- CDG-Ih
- CDG1H
- CDG syndrome type 1h
- Alpha-3-glucosyltransferase congenital disorders of glycosylation
Inheritance
ALG8-CDG is an autosomal recessive disorder meaning an affected individual inherits one defective copy of the genefrom each asymptomatic parent9.
Gene Function
The ALG8 gene encodes a glucosyltransferase enzyme, alpha-1,3-glucosyltransferase (ALG8)1,9. Glucosyltransferases are enzymes that enable the transfer of glucose molecules during glycosylation. ALG8 is located in the ER membrane where it has a role in the assembly of the LLO—a precursor for protein N-glycosylation1.The ALG8 enzyme transfers the second glucose residue from dolichol-phosphate-glucose (Dol-P-glucose) in the ER to the growing oligosaccharide chain of the LLO prior to its attachment to a protein1.
LLO synthesis
N-glycosylation is the process by which an oligosaccharide is attached to the nitrogen atom of asparagine residues on proteins. N-glycosylation is initiated in the ER and begins with the synthesis of an LLO10. The LLO is comprised of a 14-sugar oligosaccharide attached to the lipid carrier dolichol pyrophosphate (Dol-PP). The 14-sugar residues are two N-acetylglucosamine, nine mannose, and three glucose residues. Once assembled, the oligosaccharide, is transferred “en bloc” to proteins and undergoes further processing in the ER and Golgi10. Once the oligosaccharide is attached to a protein, it is referred to as an N-glycan.
LLO synthesis is carried out by a series of enzymes encoded by the DPAGT1 and ALG genes and can be divided into two phases: Phase I and Phase II (Figure 1)10.
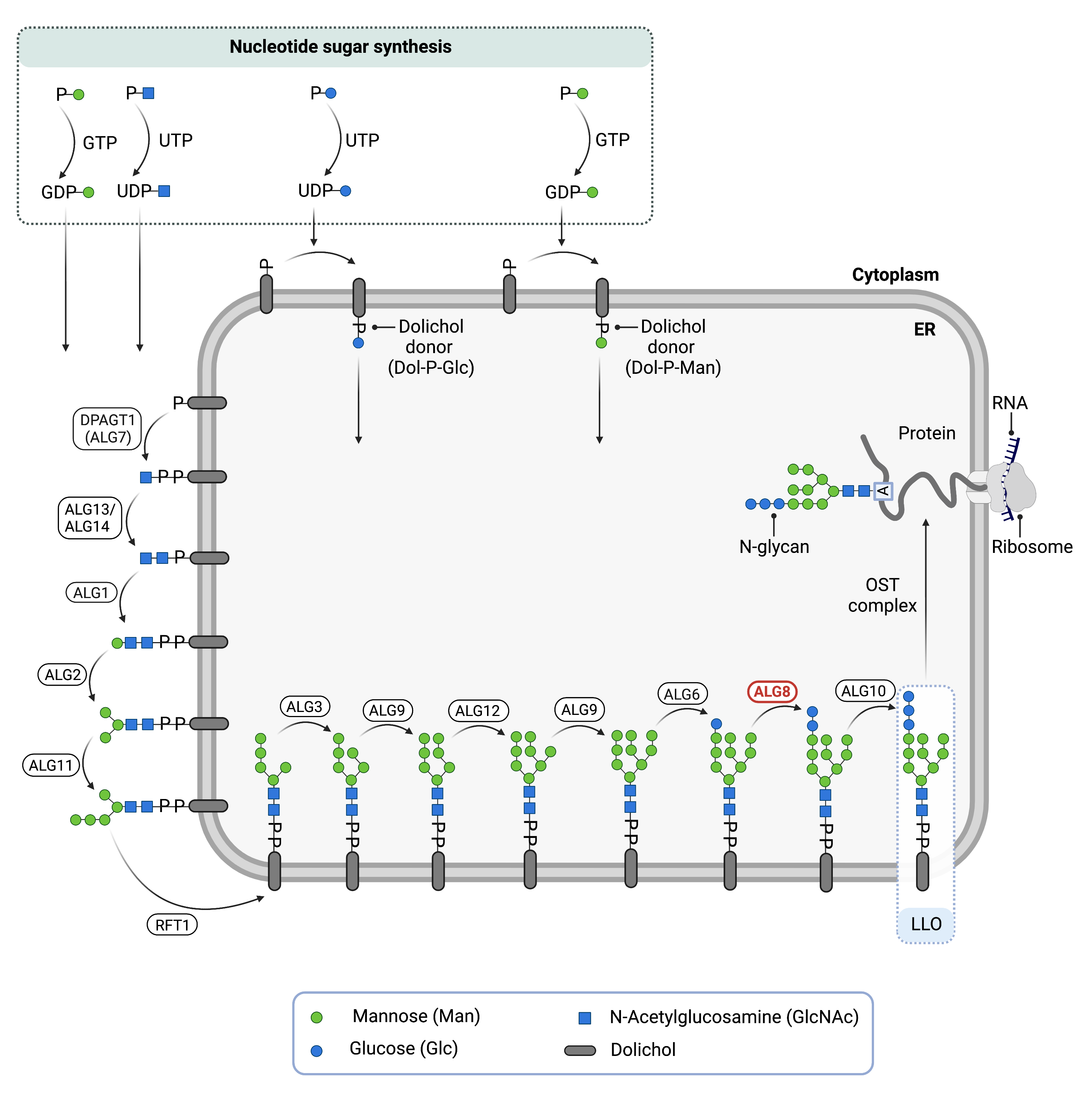
Figure 1. Role of ALG8 in glycosylation.
ALG8 is an enzyme (alpha-1,3-glucosyltransferase) involved in synthesising the lipid-linked oligosaccharide (LLO), which is needed for N-glycosylation. ALG8 adds the second glucose residue to the LLO in the endoplasmic reticulum.
Phase I
Phase I of LLO synthesis takes place on the cytoplasmic side of the ER. It begins with the sequential attachment of two N-Acetylglucosamine (GlcNAc2) and five mannose (Man5) residues to Dol-PP by several ER enzymes. GlcNAc and Man are transferred from nucleotide sugars UDP-GlcNAc and GDP-Man, respectively. The intermediate structure, Dol-PP-Man5GlcNAc2, is translocated across the ER membrane into the lumen via an enzyme by the RFT1 enzyme10,12.
Phase II
Phase II of LLO synthesis takes place in the ER lumen. Once in the lumen, four mannose residues followed by three glucose (Glc3) residues are added to the intermediate structure, generating the complete LLO, Dol-PP-Glc3Man9GlcNAc2. Mannose and glucose are transferred from glycosyl donors Dol-P-Man and Dol-P-Glc, which are formed on the cytoplasmic side of the ER and must also be flipped across the ER membrane10.
Once assembled, the oligosaccharide is transferred “en bloc” from Dol-PP to an asparagine residue of a newly synthesized protein via the enzyme oligosaccharyltransferase (OST), resulting in N-glycosylation of a protein10. The asparagine residue must be located in regions of the protein where the sequence of amino acids is asparagine-X-serine/threonine, also called Asn-X-Ser/Thr consensus sequence, where “X” can be any amino acid except proline. The activity of OST is highly specific for the completely assembled 14-sugar oligosaccharide, Glc3Man9GlcNAc2. The glucose residue added to the oligosaccharide by ALG8 is also important for proper folding and proofreading of the glycoprotein in the ER and Golgi, and improper folding leads to protein degradation10.
Disease Mechanism
Mutations in the ALG8 gene lead to the production of an abnormal enzyme with reduced or no activity. In the absence of ALG8, the LLO is missing two glucose residues. As a result, the oligosaccharide is incomplete and its transfer to the protein is impaired, causing N-glycoproteins to be under-glycosylated (hypoglycosylation)9,12.
Signs & Symptoms
Clinical Presentation
Individuals with ALG8-CDG typically develop signs and symptoms during infancy. ALG8-CDG is primarily characterized by a mild to severe neurological disorder and feeding problems. The characteristic clinical presentations of ALG8-CDG include1,9:
- Prenatal features – intrauterine growth retardation, less than expected amniotic fluid, and large fluid buildup in a baby’s tissues and organs
- Neurological – psychomotor disability, seizures, lack of coordination and muscle control (ataxia), low muscle tone (hypotonia), and structural abnormalities such as dilatation of ventricles, underdeveloped corpus callosum (hypoplasia), abnormal white matter (leukoencephalopathy/leukodystrophy), and cortical/cerebral atrophy
- Gastrointestinal – diarrhea, vomiting, feeding problems leading to failure to thrive, and protein-losing enteropathy (PLE). PLE, one of the more life-threatening symptoms, causes an excess loss of proteins in the gastrointestinal tract13
- Dysmorphic features – low-set ears, tongue swelling (macroglossia), increased distance between the eyes (hypertelorism), foot deformity (pes equnivarus), and bent and/or shortened fingers (campto- and brachydactyly)
Other symptoms may include cataracts, abnormal fat pads on arms and thighs, wrinkly skin, loose skin (cutis laxa), and inverted nipples.
Biochemical Abnormalities
Biochemical abnormalities observed in individuals with ALG8-CDG include a low blood platelet count (thrombocytopenia), elevated serum transaminases, and abnormal clotting factors1,9.
Classification
ALG8-CDG is classified as a disorder of N-linked protein glycosylation.
Under the former CDG classification system, ALG8-CDG is classified as a Type 1 CDG, which arise due to defects in the synthesis of N-glycoproteins that occur before the glycan is transferred from the LLO onto the protein.
Diagnosis
Although diagnosis of ALG8-CDG may be suspected based on presentation of symptoms and a detailed patient history, direct molecular genetic testing is the only definitive diagnostic test. Screening in suspected patients begins with a blood test to analyze serum transferrin. LLO analysis in patient fibroblasts may also be carried out following transferrin analysis1,9.
Transferrin Analysis
Individuals with ALG8-CDG show a type 1 pattern by transferrin isoelectric focusing (TIEF) or mass spectrometry analysis of transferrin. Type 1 patterns are observed in CDG that arise due to defects in LLO synthesis and are characterized by an increase in disialo- and asialo-transferrin isoforms1,9.
LLO Analysis
LLO analysis of ALG8-CDG fibroblasts show an increase in the intermediate N-glycan structure, Dol-PP-Glc1NAc2Man9, which lacks two glucose residues1,9.
Prognosis
Prognosis of ALG8-CDG may vary depending on the severity of an individual’s symptoms. Most ALG8-CDG patients are young and long-term prognosis information is unavailable. Severe forms of ALG8-CDG can lead to death in infancy and early childhood. Milder cases may show mild to severe neurological abnormalities and gastrointestinal problems (PLE). As of 2018, the oldest patient recorded in medical literature is 18 years old9.
Management
Management of symptoms may include combinations of physical therapy, occupational therapy, speech or vision therapy, and palliative measures.
Therapies
There are currently no treatment options available for ALG8-CDG. Treatment is focused on management of symptoms and prevention of complications.
Research Models
Several ALG8 research models have been generated including in yeast, fly, and mice.
Yeast (S. cerevisiae)
Alg8 mutant (YOR067C) has been generated in yeast cells. The null mutant is sensitive to reducing agents (mercaptoethanol and DTT) and shows hypoglycosylation of secreted proteins (Yeast Genome).
Fly (D. melanogaster)
Drosophila gene xiantuan (xit, CG4542, FBgn0029906) is orthologous to ALG8 and also encodes the alpha-1,3-glucosyltransferase enzyme. It is a model for the human disease ALG8-CDG (FlyBase).
Mouse (M. musculus)
Alg8 -/- knockout mouse
Heterozygous and homozygous whole body Alg8 knockout mice have been generated from the mouse line Alg8em1(IMPC)J. Homozygous mutants live from E12.5 to early adulthood and phenotypically show embryonic lethality prior to tooth bud stage (E12.5) or preweaning lethality with complete penetrance. Heterozygous mutants live to early or late adulthood and phenotypically show an increase in total amount of body fat, decreased bone mineral density and content, abnormal vibrissae morphology, decreased body length, hypoactivity, decreased lean body mass, and abnormal coat/hair pigmentation (IMPC).
Alg8 conditional-ready floxed knockout embryonic stem cell line
Embryonic stem cell line Alg8tm1a(EUCOMM)Wtsi is a targeted knockout/null mutation of Alg8. A “conditional-ready” allele can be created by flp recombinase expression in mice carrying this allele, and cre expression results in a knockout mouse. If cre is expressed without flp expression, a reported knockout mouse can be generated (MGI).
Zebrafish (D. rerio)
Ongoing research in the Ackerman lab is focused on generating a zebrafish model to understand the cellular basis of neurological defects in ALG8-CDG and to conduct drug screens (Project Grace).
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including ALG8-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Publications
ALG8-CDG Scientific Articles on PubMed
ALG8-CDG: molecular and phenotypic expansion suggests clinical management guidelines
Additional Resources
OMIM
OrphaNet
NORD
GARD
Genetic Testing Registry
ClinVar
NIH
GeneCards
UnitProt
References
- Höck, M. et al. ALG8-CDG: novel patients and review of the literature. Orphanet Journal of Rare Diseases 10, (2015).
- Chantret, I. et al. A Deficiency in Dolichyl-P-glucose:Glc1Man9GlcNAc2-PP-dolichyl α3-Glucosyltransferase Defines a New Subtype of Congenital Disorders of Glycosylation. Journal of Biological Chemistry 278, (2003).
- Bastaki, F. et al. Single‐center experience of N‐linked Congenital Disorders of Glycosylation with a Summary of Molecularly Characterized Cases in Arabs. Annals of Human Genetics 82, (2018).
- Vesela, K. et al. A new case of ALG8 deficiency (CDG Ih). Journal of Inherited Metabolic Disease 32, (2009).
- Schollen, E. Clinical and molecular features of three patients with congenital disorders of glycosylation type Ih (CDG-Ih) (ALG8 deficiency). Journal of Medical Genetics 41, (2004).
- Eklund, E. A. et al. Congenital disorder of glycosylation (CDG)-Ih patient with a severe hepato-intestinal phenotype and evolving central nervous system pathology. The Journal of Pediatrics 147, (2005).
- Stölting, T. et al. Novel ALG8 mutations expand the clinical spectrum of congenital disorder of glycosylation type Ih. Molecular Genetics and Metabolism 98, (2009).
- Charlwood, J. et al. A case of the carbohydrate-deficient glycoprotein syndrome type 1 (CDGS type 1) with normal phosphomannomutase activity. Journal of Inherited Metabolic Disease 20, (1997).
- Vuillaumier-Barrot, S. et al. Wide clinical spectrum in ALG8-CDG: clues from molecular findings suggest an explanation for a milder phenotype in the first-described patient. Pediatric Research 85, (2019).
- Bieberich, E. Synthesis, Processing, and Function of N-glycans in N-glycoproteins. in (2014). doi:10.1007/978-1-4939-1154-7_3.
- Harada, Y., Ohkawa, Y., Kizuka, Y. & Taniguchi, N. Oligosaccharyltransferase: A gatekeeper of health and tumor progression. International Journal of Molecular Sciences vol. 20 (2019).
- Stanley, P., Taniguchi, N. & Aebi, M. N-glycans. in Essentials of Glycobiology [Internet] (eds. Varki, A. et al.) (Cold Spring Harbor Laboratory Press, 2017).
- Nagra, S. & Dang, S. Protein Losing Enteropathy. in StatPearls [Internet] (StatPearls Publishing, 2021).