
Overview
Congenital disorders of glycosylation (CDG) are a group of more than 170 rare genetic disorders caused by defects in a complex chemical process known as glycosylation1. Glycosylation is a process in which enzymes act in a stepwise manner to generate, modify, and attach sugar chains, called glycans, to proteins and lipids2. Many proteins and lipids need to be glycosylated to work properly in the body and play important roles in numerous biological processes. The main glycosylation pathways in humans include N-glycosylation, O-glycosylation, glycosylphosphatidylinositol (GPI) anchor biosynthesis and lipid (glycosphingolipid) biosynthesis3,4. Glycosylation involves over 400 genes which encode various proteins and enzymes. Mutations in a number of these genes are known to cause CDG.
Since glycosylated proteins and lipids perform crucial functions in the body, defects within the glycosylation machinery can affect multiple organ systems and symptoms are often apparent from infancy5. The most common symptoms of CDG are neurological and developmental disability, including poor growth and intellectual disability6. The multi-systemic nature of CDG may also result in severe liver, gastrointestinal, and hormonal problems that require attentive care. Although a diagnosis of a CDG can be suspected based upon these symptoms, blood tests to detect abnormal glycans followed by genetic testing is necessary to confirm the specific CDG type. Currently, individual CDG types are named according the affected gene and may also be classified into five groups based on the glycosylation pathway(s) affected, which include disorders of: (i) N-linked protein glycosylation, (ii) O-linked protein glycosylation, (iii) GPI anchor biosynthesis and lipid glycosylation, (iv) multiple glycosylation pathways and (v) deglycosylation7.
There are limited therapies available for most CDG documented to date, and the high risk of severe complications and absence of a cure necessitates multi-disciplinary monitoring and management strategies for many patients7. However, multiple therapies for various CDG are in development or clinical trials, including dietary supplements, activated sugars, pharmacological chaperones, repurposed drugs, and gene therapy.
Glycosylation
Glycosylation is a complex process that attaches sugars to other molecules, including proteins and lipids. These sugar–protein and sugar–lipid complexes (also known as glycoproteins and glycolipids, respectively) are present within the cells of almost all living organisms4. Sugars can be found attached to proteins and lipids in the nucleus, cytosol, cellular compartments and cell membranes, as well as secreted substances. Given their complex, variable structures, sugars are involved in many roles in the body, such as cell attachment, signaling and immunity. As such, defects in glycosylation can lead to severe illness and decreased quality of life. Collectively, the pathologies arising from glycosylation defects are termed CDG8,4,3.
Glycosylation mostly occurs in the endoplasmic reticulum (ER) and Golgi apparatus (Golgi), although some types of glycosylation occur in the cytoplasm, nucleus and mitochondria. In humans, glycosylation can be simplified into the following steps (Figure 1):
- Cells generate sugars for glycosylation
- The sugars are converted to high-energy forms, called activated sugars, that enable them to donate their sugar component to other molecules
- These activated sugars are transported to specific locations in the cell, where enzymes transfer the sugar component to proteins, lipids, or other sugars
- Enzymes may modify the sugars attached to lipids or proteins by adding or removing more sugars or other molecules
- Often, these modified lipids or proteins are transported to a different location in the cell (such as the cell membrane).
Consequently, glycosylation requires sugars and enzymes, as well as transport systems to ensure these components are delivered to the appropriate location in the cell for each glycosylation pathway3.
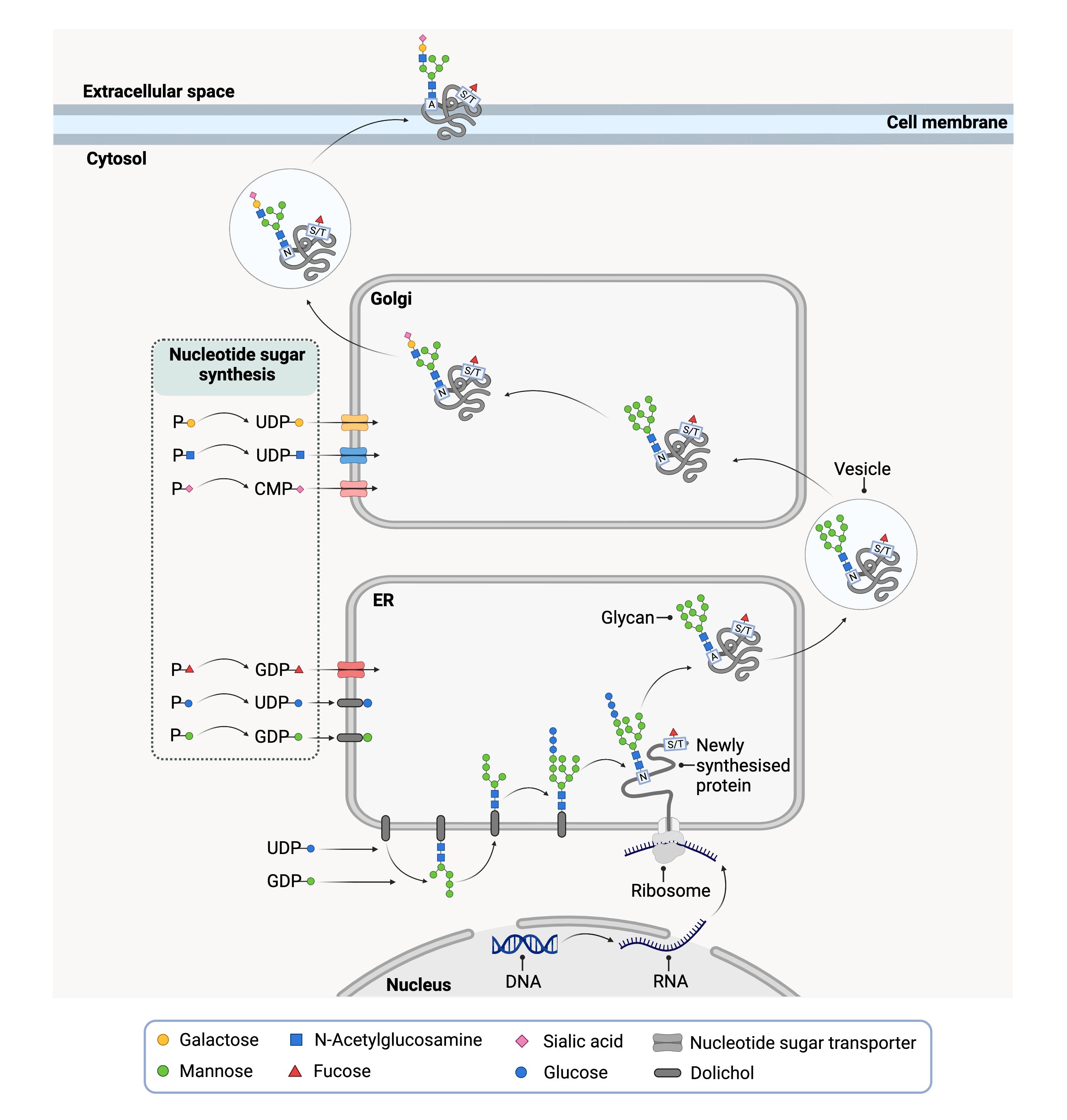
Figure 1. Overview of a simplified protein glycosylation pathway that begins in the ER.
Activated sugars are generated in the cytoplasm and transported (or flipped) into the ER where the sugar component is transferred to newly synthesized proteins. After the protein is properly folded, it is transported in a vesicle to the Golgi where additional sugars are attached. The mature protein exits the Golgi and is delivered to its final location such as the cell membrane.
Sugars and Glycans
The sugar chains generated on proteins and lipids during glycosylation are formed by simple sugars, called monosaccharides, attaching to one another. These sugar chains are also known as glycans, which can refer to any length sugar chain, from short (oligosaccharide, typically < 20 monosaccharides) to long (polysaccharide) (Figure 2) 9.
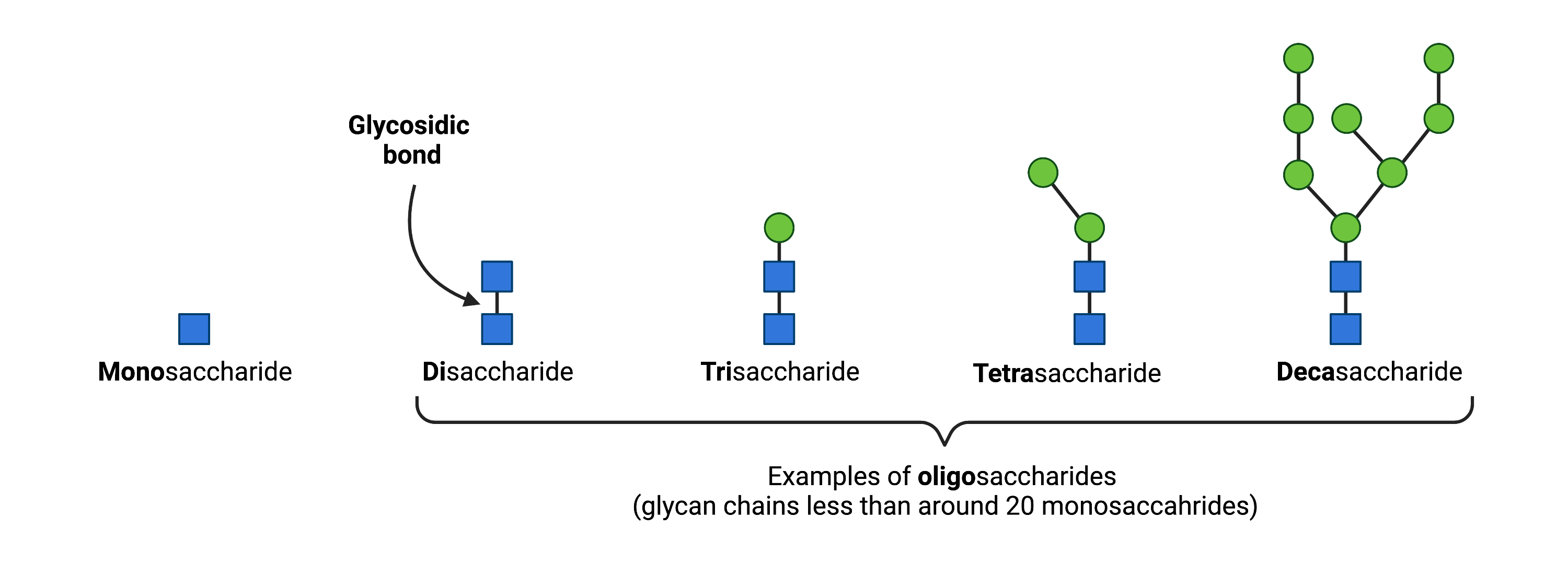
Figure 2. Oligosaccharides typically contain 2-20 monosaccharides and are classified according to their number of monosaccharide units. Monosaccharides are linked by glycosidic bonds.
In humans, the following monosaccharides can be used in glycosylation (Figure 3): glucose (Glc), galactose (Gal), N-acetyl-glucosamine (GlcNAc), N-acetyl-galactosamine (GalNAc), fucose (Fuc), glucuronic acid (GlcA), mannose (Man), N-acetylneuraminic acid (Neu5Ac), xylose (Xyl) and ribose (Rib)3,10.
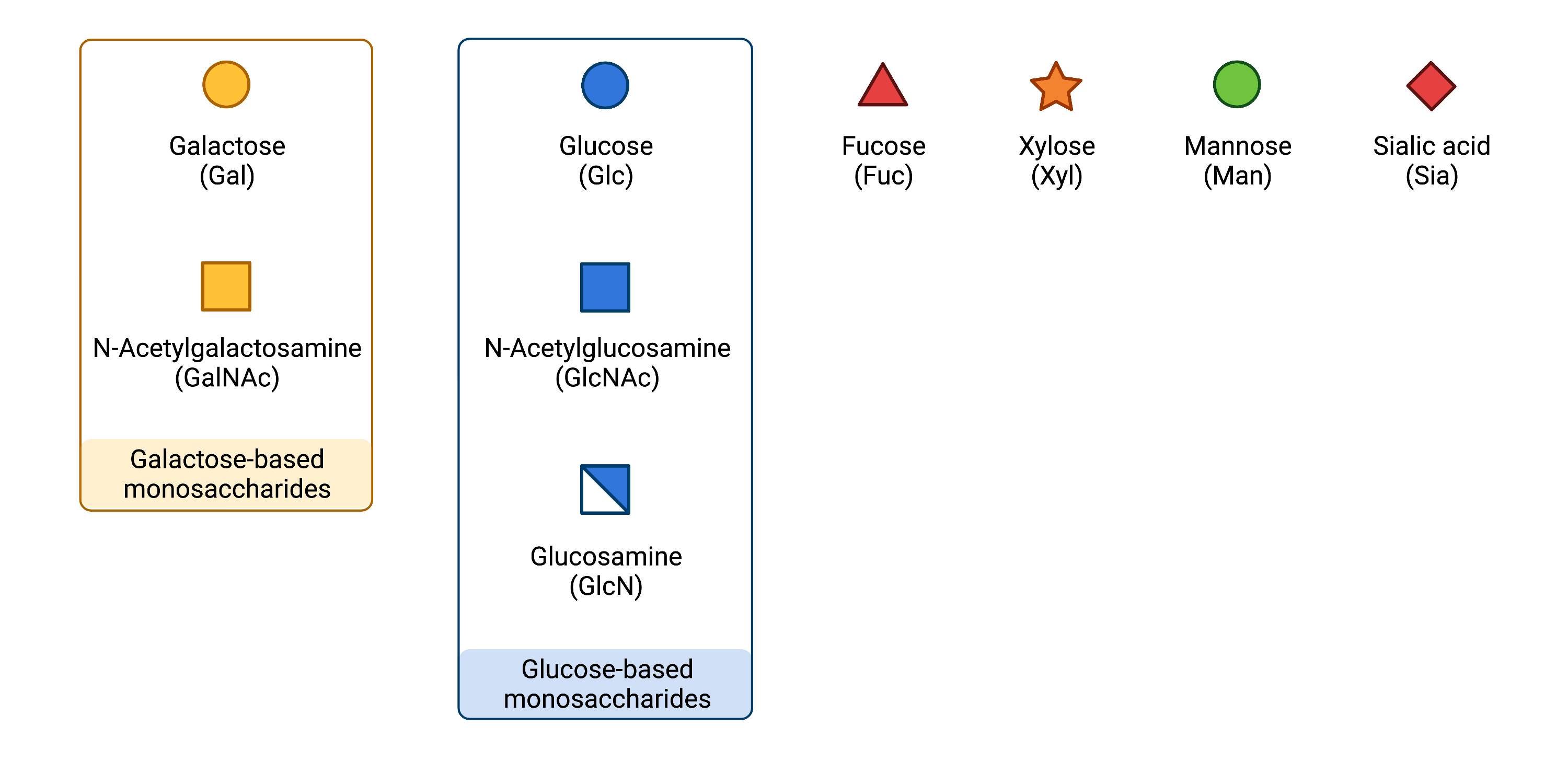
Figure 3.Common monosaccharides in glycosylation.
Monosaccharides must be converted into high-energy forms, known as activated sugars, as attaching and building glycan chains requires significant energy. Activated sugars are also called glycosyl donors since they can “donate” a sugar onto glycosyl acceptors, which may be a protein, lipid, or other sugars11. To generate activated sugars, monosaccharides are linked to phosphorylated nucleosides (such as uridine diphosphate (UDP), guanosine diphosphate (GDP) and cytidine monophosphate (CMP)) through a high-energy bond, generating nucleotide sugars (Figure 4). Breaking this high-energy bond enables the sugar component to be added to a protein, lipid, or other monosaccharide, a process that is sped-up by various glycosylation enzymes12.
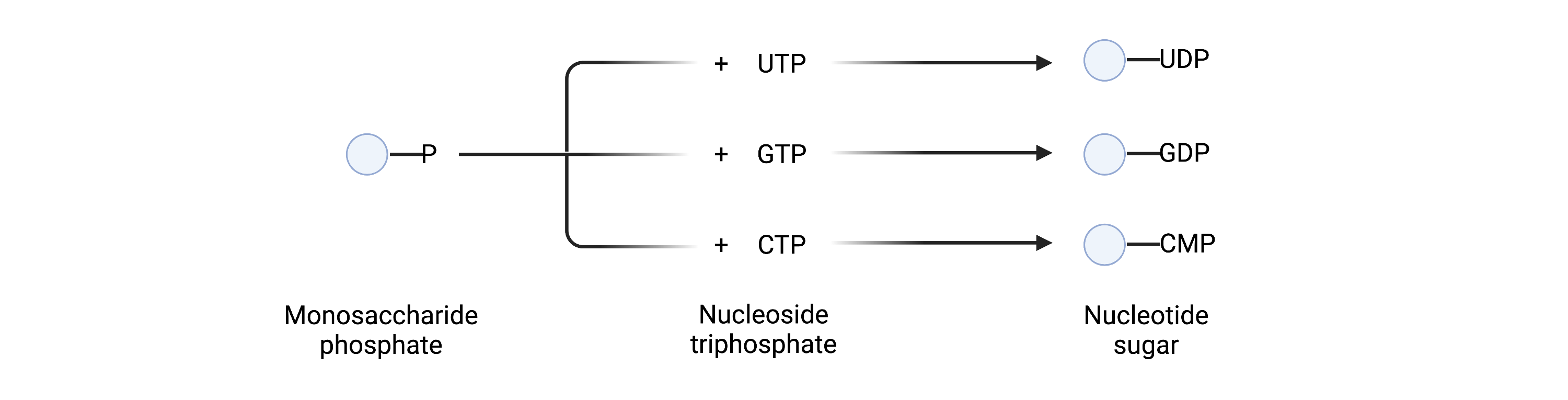
Figure 4. Synthesis of nucleotide sugars.
Phosphorylated nucleosides (UTP, GTP or CTP) are linked to monosaccharide phosphates to generate nucleotide sugars.
Nucleotide sugars can also be used to generate membrane-bound glycosyl donors by attaching their sugar to dolichol, a lipid embedded in the ER membrane, via a high-energy phosphate bond13. These dolichol-based activated sugars are important glycosyl donors inside the ER.
Glycosyltranferases
Since glycosylation is dependent on enzymes to generate, attach or modify glycan chains, many CDG are linked to defective enzymes, particularly glycosyltransferases3. Glycosyltransferases are enzymes that transfer monosaccharides (or oligosaccharides) to glycosyl acceptors, such as proteins, lipids or other glycans. Depending on where the glycosylation process is taking place, glycosyltransferases may act in the ER, Golgi, cytosol, nucleus or mitochondria3,10,14. Glycosyltransferases are selective for the saccharide they transfer and are classified by their preferred glycosyl donor, for example, glucosyltransferases add glucose residues to glycosyl acceptors15.
Sugar Transporters
Activated sugars are typically produced in the cytosol but need to be in the same location as the glycosyltransferases to be added to a protein or lipid. Glycosyltransferases can be found in the ER and Golgi, which are surrounded by membranes that activated sugars cannot pass through16. Consequently, a set of transport systems - called nucleotide sugar transporters - found in the ER and Golgi membranes, move activated sugars from the cytosol to the inside of the ER or the Golgi17. Defects in these transporters can cause major deficiencies in the glycosylation pathways, which are the cause of some CDG18.
Glycosylation Pathways
There are four main glycosylation pathways in humans:
- N-glycosylation: the process by which glycan chains are built on the nitrogen atom of an asparagine amino acid or a protein 19–21.
- O-glycosylation: the process by which glycan chains are built on the oxygen atom of certain amino acids, predominantly serine and threonine, of a protein22,23.
- GPI anchor biosynthesis: a form of glycosylation where a protein is attached to a GPI molecule, which consists of both membrane-bound lipid and glycan components; proteins attached to GPI are effectively anchored to the cell membrane24,25.
- Lipid glycosylation (glycosphingolipid biosynthesis): a form of lipid glycosylation where glycans are attached to ceramide (a specific sphingolipid), forming glycosphingolipids26.
Within these pathways, glycosylation can be further classified based on other factors, such as the first monosaccharide in a glycan chain to be attached to a protein or lipid3,4. CDG are broadly classified based on which glycosylation pathway(s) is most affected, e.g., a disease that arises due to a defect in N-glycosylation is known as an N-linked CDG.
CDG Classification
Type I & Type II
Traditionally, CDG were classified into two groups (Type I/CDG-I or Type II/CDG-II) depending on whether the glycosylation defect occurred before or after the transfer of the oligosaccharide to a protein. However, this classification system could only differentiate CDG caused by defects in N-glycosylation and is no longer used to classify CDG types8.
- Type I/CDG-I: caused by defects in the assembly and transfer of oligosaccharides to an asparagine residue on a protein
- Type I/CDG-II: caused by defects in the processing of the N-glycans attached to proteins
Individual CDG Types
As more CDG were identified, the “two-type” classification system became too complex and inconsistent, and CDG caused by defects in different or multiple glycosylation pathways could not be classified by this method27. As of 2009, the nomenclature used to classify CDG includes the name of the defective gene, followed by “-CDG” (e.g. PMM2-CDG)28.
Glycosylation Pathways
CDG may be broadly classified into five categories depending on the glycosylation pathway(s) affected, including disorders of N-linked glycosylation, disorders of O-Linked protein glycosylation, disorders of lipid and GPI anchor biosynthesis, disorders of multiple glycosylation pathways and disorders of deglycosylation.
- Disorders of N-linked Protein Glycosylation: arise from defects within the N-glycosylation pathway, including the assembly, attachment, and processing of N-glycan chains.
- Disorders of O-linked Protein Glycosylation: arise from defects within the O-glycosylation pathway, including in the generation, attachment, and modification of O-glycan chains.
- Disorders of Lipid Glycosylation and GPI Anchor Biosynthesis: disorders of GPI anchor biosynthesis are caused by defects in the synthesis and attachment of GPI anchors to proteins, and disorders of lipid glycosylation are caused by defects in the synthesis of a subtype of lipids called glycosphingolipids.
- Disorders of Multiple Glycosylation Pathways: are caused by defects that affect more than one glycosylation pathway.
- Disorders of Deglycosylation: are caused be defects in the removal defects in the removal of N-glycans from proteins and their subsequent breakdown. Also called congenital disorders of deglycosylation (CDDG).29-31
Signs & Symptoms
Given the ubiquitous presence of glycosylation pathways in the body, CDG may affect multiple organ systems although most cases involve neurological abnormalities. The clinical presentation of CDG varies widely both within and between types and symptoms may range from mild to severe, even among individuals with the same CDG type. Most CDG present with multi-systemic involvement in infancy, although the onset of symptoms may occur in adulthood for some CDG types. The complete clinical spectrum is not well characterized for many CDG types because few affected individuals have been reported5.
Symptoms in infants and children may include 5,32,33:
- Growth problems –feeding difficulties, failure to thrive
- Development delays – intellectual disability, delay in reaching developmental milestones
- Neurological problems – decreased muscle tone (hypotonia), seizures, poor balance and coordination (ataxia), cerebellar abnormalities, stroke-like episodes
- Ophthalmological problems – misaligned or crossed eyes (strabismus), eye abnormalities, retinal degeneration (retinitis pigmentosa)
- Liver problems – liver disease with elevated liver enzymes
- Heart problems – fluid accumulation around the heart, thickening and stiffening of the heart muscle (cardiomyopathy)
- Hematologic problems – abnormal bleeding or blot clotting
- Endocrine problems – low blood sugar due to high insulin levels (hypoglycemia), decreased thyroid hormone (hypothyroidism), growth hormone deficiency
- Skeletal problems – curving of the spine
- Immunologic problems – severe or long-lasting infections
- Gastrointestinal symptoms - chronic diarrhea, vomiting
- Skin problems - loose skin; rash, scaly skin, peeling of skin
Symptoms that may develop in adulthood include:
- Intellectual disability
- Ataxia
- Slurred speech (dysarthria)
- No puberty in girls
- Joint problems
- Liver fibrosis
- Retinitis pigmentosa
Diagnosis
CDG are difficult to diagnose as severity and symptoms can vary between cases34. Upon clinical suspicion, there are a series of tests that may be performed to diagnose an individual with a CDG and determine CDG type. These tests are typically performed in a specific order, as results from one test inform the selection of the appropriate subsequent tests to help reach a diagnosis (Figure 5).
Common CDG diagnostic tests include:
- Glycan analysis
- Enzymatic analysis
- LLO analysis
- Genetic testing
Clinicians can order initial screening tests for individuals presenting CDG symptoms, which are typically blood tests that look for abnormal glycan patterns on certain proteins (such as transferrin and apolipoprotein C-III). These initial screening tests can help identify which glycosylation pathway is affected35.
As glycosylation is a process that relies heavily on enzymes, CDG can arise from defective enzymes36. Abnormal glycan patterns identified in the initial screening tests may indicate defects in a particular enzyme. Further tests may then be performed to measure the activity of suspected defective enzymes, which can identify specific CDG type37. But for most CDG cases, molecular genetic testing is the only definitive method for diagnosis38.
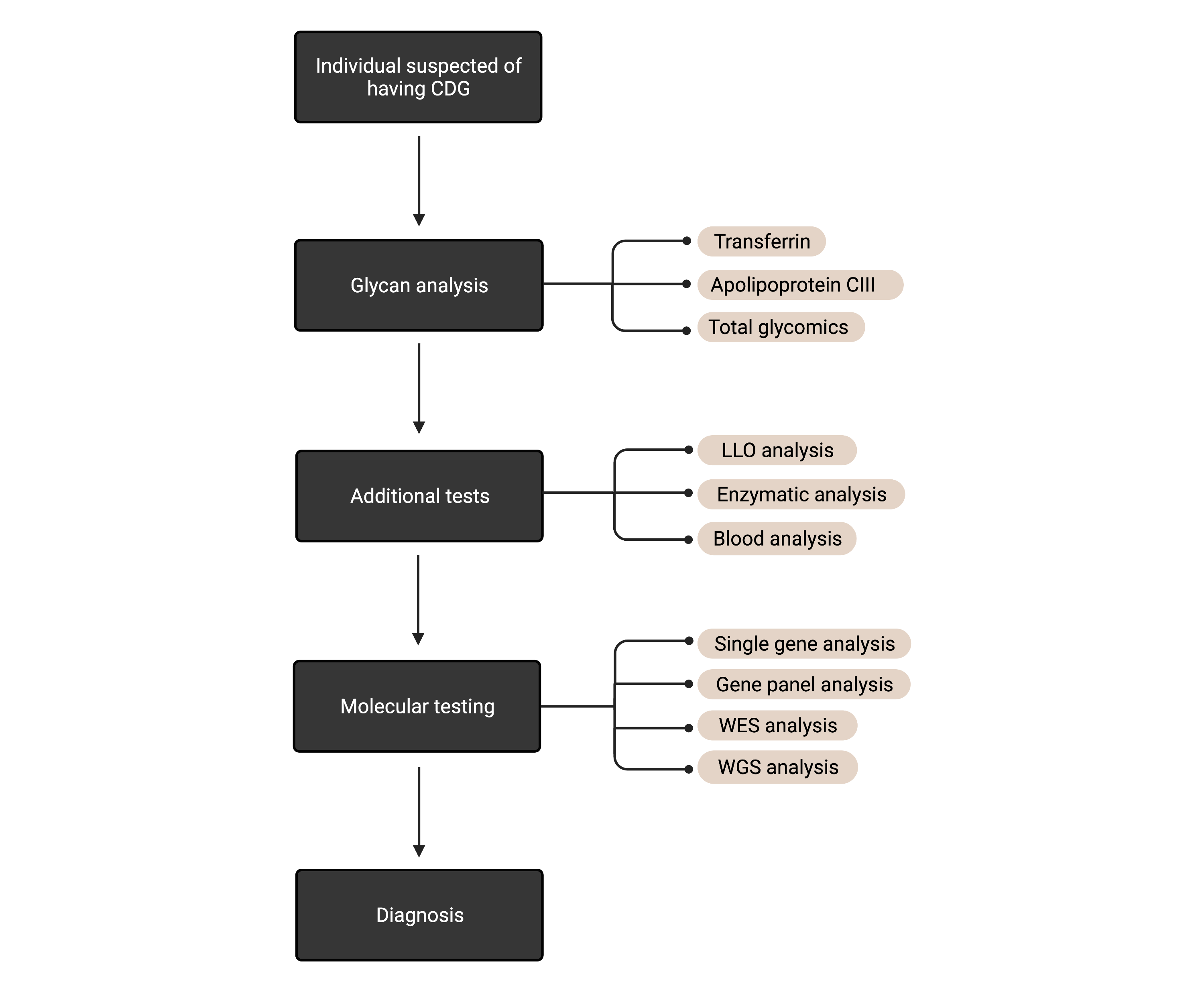
Figure 5. The process for diagnosing suspected cases of CDG.
First-line screening tests typically include blood tests that look for abnormal glycan patterns on specific serum proteins (transferrin, apolipoprotein CIII) or total glycans. Subsequent tests may include enzyme analysis or various blood tests. For most CDGs, genetic testing is required for a definitive diagnosis.
Glycan Analysis
Initial testing for suspected cases of CDG are usually blood tests that detect abnormal glycan patterns on proteins; proteins with different glycosylation patterns are known as isoforms. Two proteins commonly analyzed in CDG screening are transferrin and apolipoprotein C-III. Combining results from transferrin and/or apolipoprotein C-III tests with clinical features can inform clinicians of the most appropriate follow-up strategy, such as enzyme or molecular testing37,38.
Transferrin
Transferrin analysis, also called transferrin isoform analysis or carbohydrate deficient transferrin (CDT) analysis, is a first-line screening test for assessing suspected CDG cases39. Transferrin is an abundant glycoprotein found in the blood that transports iron around the body40. Transferrin has two branching N-linked glycan chains that are each terminated with the monosaccharide: sialic acid41. The glycan chains can be terminated with different numbers of sialic acid residues, generating different transferrin isoforms or glycoforms.
Transferrin isoforms are named by the number of sialic acid residues found on the glycan chains, such as asialosialo-, monosialo-, disialo-, trisialo-, and tetrasialo-, pentasialo- or hexasialo-transferrin, which contain zero, one, two, three, four, five or six sialic acid residues, respectively (Figure 6) 42. The relative levels of these different transferrin isoforms (or ‘transferrin patterns’) can help detect the presence of CDG. However, because transferrin only has N-glycans, CDT analysis can only be used to detect CDG caused by N-glycosylation defects28.
Figure 6. Serum Transferrin Isoforms.
A. Transferrin contains two N-glycans and B. has different isoforms (glycoforms) depending on the number of sialic acid residues present on the glycans. Transferrin isoforms are named according to the number of sialic acid residues present. Tetrasialo-transferrin is the most abundant isoform in healthy individuals.
In healthy individuals, tetrasialo-transferrin is typically the most abundant isoform detected. In contrast, individuals with CDG often have abnormal transferrin patterns, displaying higher levels of isoforms that have fewer sialic acid residues or are missing complete glycan chains. Transferrin isoforms that lack sialic acid residues are referred to as carbohydrate deficient transferrin (CDT)43. However, in some CDG cases, abnormal transferrin patterns are not detected or can normalize over time44.
There are two types of abnormal transferrin patterns in CDG cases, named Type I and Type II (Figure 7):
- Type I pattern: characterized by decreased levels of tetrasialo-transferrin and increased levels of asialo- and/or disialo-transferrin isoforms44. A type 1 pattern is often associated with defects in the assembly or transfer of the glycan from the LLO to a protein45,46.
- Type II pattern: characterized by increased levels in asialo-, monosialo-, disialo-, and/or trisialo-transferrin44. A type 2 pattern is often associated with defects that occur after glycan transfer to the protein in the ER46.
CDG were traditionally classified based on these transferrin patterns, as either CDG-I or CDG-II, followed by a letter (such as CDG-IIa) to denote a different CDG type. As this classification system presented inconsistencies and limited to defects associated with N-glycans, CDG are now typically classified by the defective gene (such as MGAT2-CDG)28.
The specific method performed for CDG transferrin screening tests often depends on the instruments available in the diagnostic lab and can include isoelectric focusing (IEF), mass spectrometry, high-pressure liquid chromatography (HPLC) and capillary electrophoresis55,57.
Transferrin Isoelectric Focusing (TIEF)
One of the most frequently used screening methods for transferrin glycan abnormalities is transferrin isoelectric focusing (TIEF), where transferrin isoforms are separated based on the pH at which an isoform has an overall neutral charge (known as the “isoelectric point”). The more sialic acids present on transferrin, the lower the isoelectric point value and the closer it appears near the top of the gel. Depending on technique, individual isoforms can be detected during analysis, which can be used to observe transferrin patterns. For example, traditional IEF relies on gels where individual isoforms are displayed as different bands on the gel (Figure 7)47.
Figure 7. Example of transferrin isoelectric focusing results for healthy and N-linked CDG samples.
Compared to healthy samples (left), CDG Type I CDG (middle) are characterized by increased levels of asialo- and diasialo-transferrin and Type II CDG (right) are characterized by increased levels of asialo-, monosialo-, disialo-, and/or trisialo-transferrin, as indicated by darker bands on the gel.
Mass Spectometry
Mass spectrometry is often used to analyze serum transferrin, either in addition to or in place of TIEF48–51. This type of analysis can identify the exact sugars present in the glycan chain, which can serve as molecular fingerprints for some CDG types.
Apolipoprotein C-III
Analysis of the glycoprotein apolipoprotein C-III is used to diagnose certain O-linked CDG. As well, if a type II pattern is observed in transferrin analysis, analysis of apolipoprotein C-III is carried out because some type II patterns can result from defects in Golgi homeostasis, which also affects O-glycosylation52. Apolipoprotein C-III is a glycoprotein that is also found in the blood, where it helps to control triglyceride-rich lipoprotein levels53. Apolipoprotein C-III has a single O-glycan attached to a threonine residue that can adopt different isoforms with different numbers of sialic acid residues (Figure 8). These isoforms can including zero/asialo (apo-CIII0), one/monosialo (apoC-III1) or two/disialo (apoC-III2) sialic acid residues54.
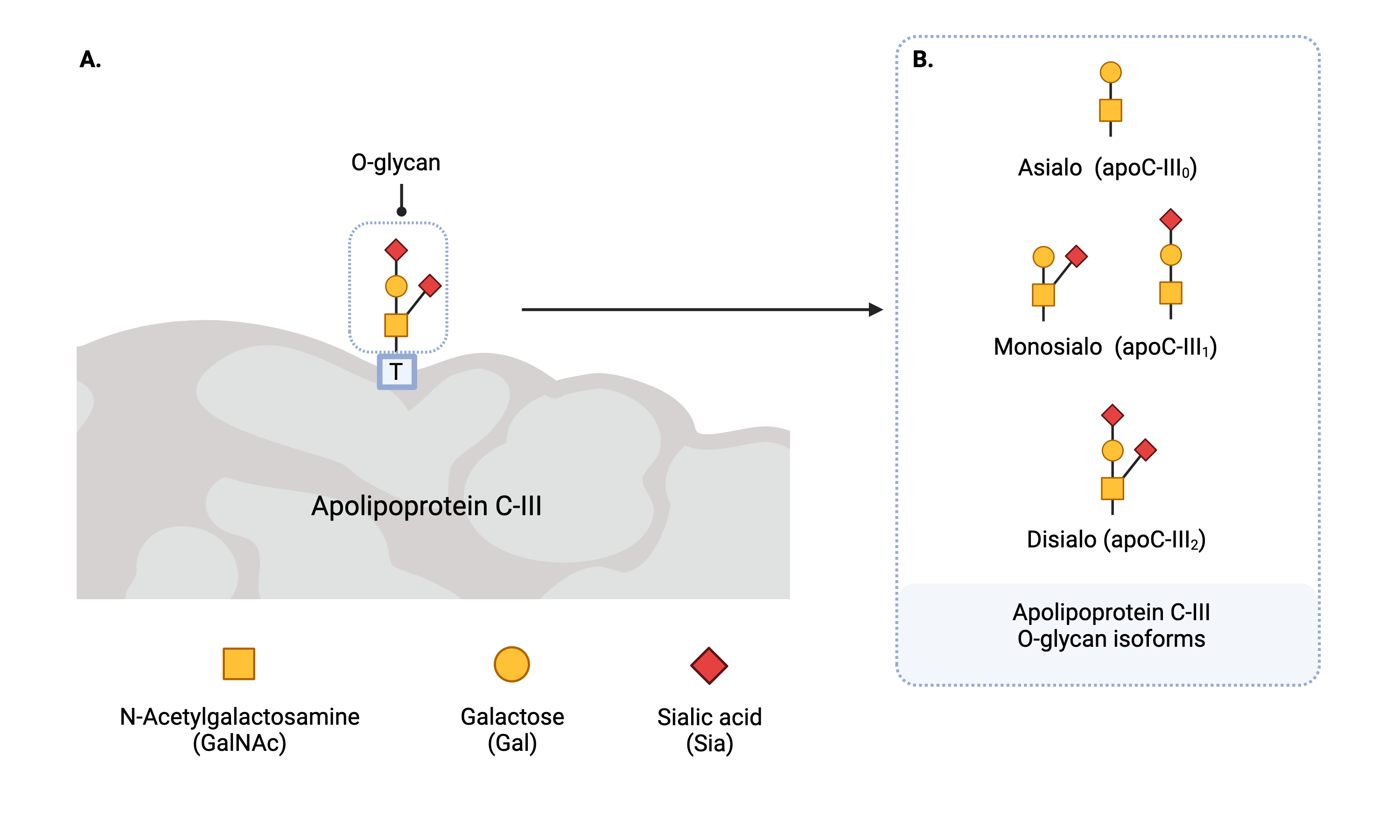
Figure 8. Apolipoprotein C-III isoforms.
Apolipoprotein CIII (Apo C-III) contains a single O-glycan and is present in different isoforms (glycoforms) depending the number of sialic acid residues present on the glycan. Apo C-III isoforms are named according to the number of sialic acid residues present.
Apolipoprotein C-III screening can detect CDG that arise due to defects involving a type O-glycosylation called O-GalNAcylation and defects in Golgi homeostasis by increased levels of apo C-III0 and/or apo C-III1 (Figure 9) 52. Like transferrin analysis, serum apolipoprotein CIII can be screened by various laboratory methods including isoelectric focusing, two-dimensional gel electrophoresis, capillary electrophoresis and mass spectrometry52,55.
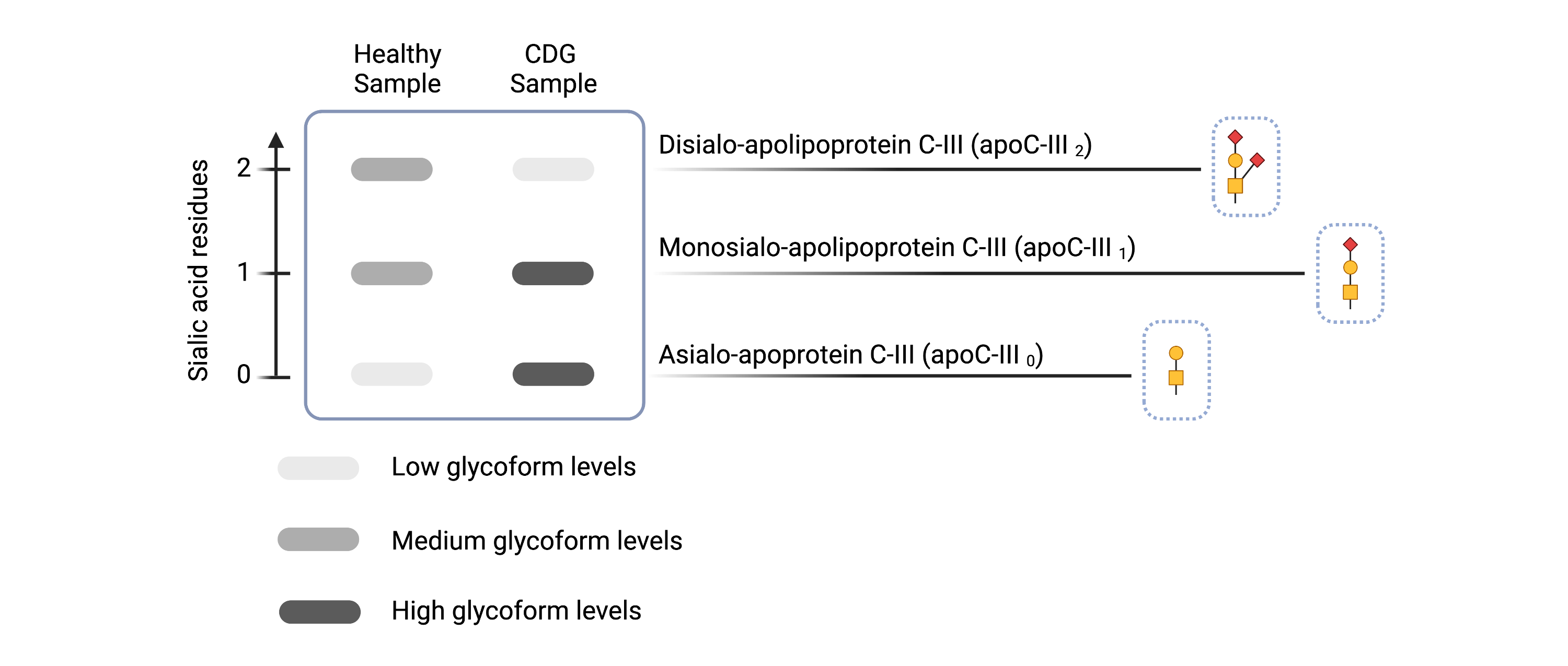
Figure 9. Example of Apolipoprotein C-III isoelectric focusing (IEF) results for healthy and CDG samples. Apolipoprotein C-III IEF can detect some O-linked CDGs and CDGs that involve Golgi defects.
Total Glycans Analysis
In addition to analyzing the glycans on transferrin (N-glycans) and apolipoprotein C-III (O-glycan) for abnormalities, all glycans (or total glycans) in a patient sample can be analyzed by mass spectrometry. This diagnostic method is called glycan profiling or glycoprofiling and is often performed on serum or plasma samples collected from patients. In this method, samples are first enzymatically or chemically treated in order to cleave all glycans that are attached to proteins. Using mass spectrometry, total N-glycans and/or total O-glycans can be analyzed for abnormal patterns that may be suggestive of CDG. Although blood samples are typically analyzed, glycoprofiling may also be performed on urine or cerebrospinal fluid samples collected from patients48,56.
Enzyme Analysis
Some CDG that display a type I transferrin pattern may be investigated for mutations in genes encoding specific enzymes, such as PMM2 and MPI42. Generally, these analyses are carried out in fibroblast or leukocyte cells, where suspected enzymes are extracted from skin or blood samples and enzyme activity is measured37. If enzyme activities are normal, additional analyses on PMM2 and MPI may be required, such as LLO analysis57,58.
LLO Analysis
LLO analysis can detect type I CDG that are caused by defects in the synthesis or transfer of the LLO, such as ALG8-CDG, RFT1-CDG and ALG1-CDG42,59. In LLO analysis, fibroblasts are collected from skin samples, specific monosaccharides are radiolabeled and the oligosaccharide component of the LLO is analysed for abnormal composition such as absent monosaccharides57.
Genetic Testing
Molecular testing can provide definitive diagnoses by identifying defective genes in CDG cases, using assessments such as gene panels, whole-exome sequencing (WES) and whole-genome sequencing (WGS)37,60. Gene panels are genetic tests that screen multiple genes to identify any mutations present in these genes. More extensive genetic screening can be performed if gene panel analysis does not identify a mutation in any suspected genes. WES is used to assess for any mutations in coding regions of DNA, and WGS is used to assess both coding and noncoding regions of DNA61,36.
Management
Treatment for most CDG types is primarily supportive and is directed towards managing the specific symptoms that are present in each individual and preventing complications. Management of CDG depends on the CDG type, which body systems are affected, symptoms, age, and other health conditions. Multidisciplinary care is often required and may include
occupational, speech, and physical therapy, as well as management of neurological, endocrinological, hematological and gastroenterological symptoms by specialists5.
Standard care for CDG may be focused on:
- Monitoring the function of organ and body systems – laboratory tests to monitor liver, thyroid, kidney, and endocrine function, coagulation and vision
- Rehabilitation - occupational, speech and physical therapy for developmental delays and balance and coordination problems
- Feeding therapy - for growth and eating problems, including the use of special formulas to maximize caloric intake, thickening of liquids, and/or the use of feeding tubes
- Blood clot prevention – individuals with CDG are at a higher risk of blood clots and may require blood-thinner medications or plasma infusions
- Treating misaligned or crossed eyes – corrective lenses, patching or surgery to treat eye problems
- Thyroid problems – symptoms such as slow weight gain, growth delay and low blood sugar may arise due to thyroid problems and may be treated with hormone medication
- Seizure management – anti-epileptic medications, hormones, dietary supplements, diet changes or epilepsy surgery to manage different types of seizures
- Life skills and vocational training – enabling independent living of adolescents and adults with CDG
Therapies
Specific treatments are available for several CDG which mainly consist of dietary sugar supplementation and in some cases, organ transplantation. Other treatments including repurposed drugs, pharmacological chaperones, and activated sugars, have shown to improve clinical symptoms in some CDG and are currently under investigation in clinical trials.
Specific treatments are available or are under clinical investigation for the following CDG1,7,62:
Dietary Supplementation
Dietary supplementation is one of the most widely applied treatment strategies for CDG. The goal of dietary supplementation is to provide specific monosaccharides, such as the substrate or product of the affected enzyme, to bypass this step of the pathway and restore normal glycosylation7,63.
Organ Transplantation
Organ transplantation has been a successful treatment option in cases with severe and progressive heart or liver involvement. Organ transplantation has been shown to relieve some symptoms and cure certain CDG1,64–68.
Repurposed Drugs
Drug repurposing involves re-using an existing drug to treat new diseases. Several repurposed drugs are currently under investigation for the treatment of PMM2-CDG. Acetazolamide, a medication that is used to treat glaucoma, intracranial hypertension and epilepsy, has been shown to improve neurological symptoms in PMM2-CDG patients69 and is currently under investigation in a clinical trial to determine its efficacy in approving ataxia in PMM2-CDG patients64. The aldose reductase inhibitor epalrestat which is used to treat sorbitol-related neuropathies, is also under investigation for its ability to restore some enzymatic activity in PMM2-CDG patients1.
Pharmacological Chaperones
Pharmacological chaperones are small molecules that bind directly to the mutant protein and restore its function by ensuring its proper folding and stability. This therapeutic approach is currently under investigation to treat PMM2-CDG70.
Activated Sugars
Monosaccharides must be “activated” to a high-energy form to be used as donors in glycosylation. The activated sugar mannose-1-phosphate is currently in development as a therapy for PMM2-CDG71.
Gene Therapy
Gene therapy typically involves replacing a disease-causing gene with a healthy copy of a gene to treat or cure disease. Gene therapy is currently being investigated to treat several types of CDG including SRD5A3-CDG and PIGA-CDG72.
Prognosis
The prognosis of CDG varies between different types of CDG, as well as between individuals with the same CDG type7. Therefore, life expectancy cannot be predicted for patients without further clinical information5.
However, a 25% mortality rate has been identified for young children diagnosed with CDG. This has been correlated to complications resulting from multi-systemic illness during early childhood. Accordingly, children presenting without such complications during their first years may lead steady lives, with greater management focus directed to their growth and development. With improving awareness about CDG, resulting in increased diagnosis rates and better management through multi-disciplinary teams, children diagnosed with CDG are able to lead fulfilling and meaningful lives5.
Epidemiology
The incidence and prevalence of CDG are not well established, but are thought to remain largely under-diagnosed36. Individuals with CDG have been reported worldwide from most ethnic backgrounds. The prevalence of CDG in European populations has been estimated to range from 1/10,000 to 0.1–0.5/100,00036. There are limited reports of CDG prevalence within specific countries or populations.
CDG affect both sexes similarly, as most CDG are inherited in an autosomal recessive manner. Exceptions include POGLUT1-CDG, POFUT1-CDG, EXT1-CDG, EXT2-CDG, GANAB-CDG, SEC63-CDG and PRKCSH-CDG, which are inherited in an autosomal dominant manner. Other exceptions include CDG that are inherited in a X-linked manner, such as ATP6AP1-CDG, C1GALT1C1-CDG, PIGA-CDG, SSR4-CDG, SLC35A2-CDG, ALG13-CDG and MAGT1-CDG5.
Review Publications
References
- Ondruskova, N., Cechova, A., Hansikova, H., Honzik, T. & Jaeken, J. Congenital disorders of glycosylation: Still “hot” in 2020. Biochimica et Biophysica Acta - General Subjects vol. 1865 (2021).
- Apweiler, R., Hermjakob, H. & Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta - Gen. Subj. 1473, (1999).
- Schjoldager, K. T., Narimatsu, Y., Joshi, H. J. & Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. doi:10.1038/s41580-020-00294-x.
- Reily, C., Stewart, T. J., Renfrow, M. B. & Novak, J. Glycosylation in health and disease. Nature Reviews Nephrology vol. 15 346–366 (2019).
- Lee, Y. C., Brubaker, P. L. & Drucker, D. J. Developmental and tissue-specific regulation of proglucagon gene expression. Endocrinology 127, 2217–2222 (1990).
- Bullock, B. P., Heller, R. S. & Habener, J. F. Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology 137, 2968–2978 (1996).
- Verheijen, J., Tahata, S., Kozicz, T., Witters, P. & Morava, E. Therapeutic approaches in Congenital Disorders of Glycosylation (CDG) involving N-linked glycosylation: an update. Genetics in Medicine vol. 22 (2020).
- Lekka, D. E. et al. Congenital disorders of glycosylation – an umbrella term for rapidly expanding group of rare genetic metabolic disorders – importance of physical investigation. Bratislava Med. J. 122, (2021).
- Seeberger, P. H. Chapter 2 Monosaccharide Diversity. Essentials Glycobiol. 3rd Ed. (2017).
- Freeze, H. H., Chong, J. X., Bamshad, M. J. & Ng, B. G. Solving glycosylation disorders: Fundamental approaches reveal complicated pathways. American Journal of Human Genetics vol. 94 (2014).
- Varki, A. et al. Essentials of glycobiology, third edition. Cold Spring Harbor Laboratory Press (2017).
- Mikkola, S. Nucleotide sugars in chemistry and biology. Molecules vol. 25 (2020).
- Rini, J. M. & Esko, J. D. Glycosyltransferases and Glycan-Processing Enzymes - Essentials of Glycobiology - 3rd Edition. in Essentials of Glycobiology (2017).
- Joshi, H. J. et al. Glycosyltransferase genes that cause monogenic congenital disorders of glycosylation are distinct from glycosyltransferase genes associated with complex diseases. Glycobiology 28, (2018).
- Biswas, A. & Thattai, M. Promiscuity and specificity of eukaryotic glycosyltransferases. Biochemical Society Transactions vol. 48 (2020).
- Niemann, M. C. & Werner, T. Endoplasmic reticulum: Where nucleotide sugar transport meets cytokinin control mechanisms. Plant Signal. Behav. 10, (2015).
- Hadley, B. et al. Structure and function of nucleotide sugar transporters: Current progress. Computational and Structural Biotechnology Journal vol. 10 (2014).
- Parker, J. L. & Newstead, S. Structural basis of nucleotide sugar transport across the Golgi membrane. Nature 551, (2017).
- Breitling, J. & Aebi, M. N-linked protein glycosylation in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 5, (2013).
- Lee, H. S., Qi, Y. & Im, W. Effects of N-glycosylation on protein conformation and dynamics: Protein Data Bank analysis and molecular dynamics simulation study. Sci. Rep. 5, (2015).
- Wild, R. et al. Structure of the yeast oligosaccharyltransferase complex gives insight into eukaryotic N-glycosylation. Science (80-. ). 359, (2018).
- van Tol, W., Wessels, H. & Lefeber, D. J. O-glycosylation disorders pave the road for understanding the complex human O-glycosylation machinery. Current Opinion in Structural Biology vol. 56 (2019).
- Christlet, T. H. T. & Veluraja, K. Database analysis of O-glycosylation sites in proteins. Biophys. J. 80, (2001).
- Kinoshita, T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biology vol. 10 (2020).
- Kinoshita, T. Glycosylphosphatidylinositol (GPI) anchors: Biochemistry and cell biology: Introduction to a thematic review series. Journal of Lipid Research vol. 57 (2016).
- MacCioni, H. J. F., Quiroga, R. & Ferrari, M. L. Cellular and molecular biology of glycosphingolipid glycosylation. Journal of Neurochemistry vol. 117 (2011).
- Jaeken, J., Hennet, T., Freeze, H. H. & Matthijs, G. On the nomenclature of congenital disorders of glycosylation (CDG). J. Inherit. Metab. Dis. 31, (2008).
- Jaeken, J., Hennet, T., Matthijs, G. & Freeze, H. H. CDG nomenclature: Time for a change! Biochim. Biophys. Acta - Mol. Basis Dis. 1792, (2009).
- Lipari Pinto, P. et al. Ngly1 deficiency—a rare congenital disorder of deglycosylation. JIMD Rep. 53, (2020).
- Enns, G. M. et al. Mutations in NGLY1 cause an inherited disorder of the endoplasmic reticulum-associated degradation pathway. Genet. Med. 16, (2014).
- Lam, C. et al. Prospective phenotyping of NGLY1-CDDG, the first congenital disorder of deglycosylation. Genet. Med. 19, (2017).
- Marques-da-Silva, D. et al. Liver involvement in congenital disorders of glycosylation (CDG). A systematic review of the literature. Journal of Inherited Metabolic Disease vol. 40 (2017).
- Marques-da-Silva, D. et al. Cardiac complications of congenital disorders of glycosylation (CDG): a systematic review of the literature. Journal of Inherited Metabolic Disease vol. 40 (2017).
- Medrano, C. et al. Clinical and molecular diagnosis of non-phosphomannomutase 2 N-linked congenital disorders of glycosylation in Spain. Clin. Genet. 95, (2019).
- Heywood, W. E. et al. Global serum glycoform profiling for the investigation of dystroglycanopathies & Congenital Disorders of Glycosylation. Mol. Genet. Metab. Reports 7, (2016).
- Péanne, R. et al. Congenital disorders of glycosylation (CDG): Quo vadis? European Journal of Medical Genetics vol. 61 (2018).
- Čechová, A. et al. Consensus guideline for the diagnosis and management of mannose phosphate isomerase-congenital disorder of glycosylation. Journal of Inherited Metabolic Disease vol. 43 (2020).
- Calvo, P. L. et al. An unexplained congenital disorder of glycosylation-II in a child with neurohepatic involvement, hypercholesterolemia and hypoceruloplasminemia. in JIMD Reports vol. 38 (2018).
- Casetta, B. et al. A new strategy implementing mass spectrometry in the diagnosis of congenital disorders of N-glycosylation (CDG). Clin. Chem. Lab. Med. 59, (2020).
- Ogun, A. S. & Adeyinka, A. Biochemistry, Transferrin. StatPearls (2018).
- Wada, Y. Mass spectrometry of transferrin glycoforms to detect congenital disorders of glycosylation: Site-specific profiles and pitfalls. Proteomics 16, (2016).
- Goreta, S. S., Dabelic, S. & Dumic, J. Insights into complexity of congenital disorders of glycosylation. Biochemia Medica vol. 22 (2012).
- Dave, M. B. et al. Comparison of transferrin isoform analysis by capillary electrophoresis and HPLC for screening congenital disorders of glycosylation. J. Clin. Lab. Anal. 32, (2018).
- Lefeber, D. J., Morava, E. & Jaeken, J. How to find and diagnose a CDG due to defective N-glycosylation. Journal of Inherited Metabolic Disease vol. 34 (2011).
- Asteggiano, C. G. et al. Ten years of screening for congenital disorders of glycosylation in Argentina: case studies and pitfalls. Pediatr. Res. 84, (2018).
- de Magalhães, A. P. P. S. et al. Transferrin isoelectric focusing for the investigation of congenital disorders of glycosylation: analysis of a ten-year experience in a Brazilian center. J. Pediatr. (Rio. J). 96, (2020).
- Wu, J., Haitjema, C. H., Heger, C. D. & Boge, A. A proof-of-concept analysis of carbohydrate-deficient transferrin by imaged capillary isoelectric focusing and in-capillary immunodetection. Biotechniques 68, (2020).
- Abu Bakar, N., Lefeber, D. J. & van Scherpenzeel, M. Clinical glycomics for the diagnosis of congenital disorders of glycosylation. J. Inherit. Metab. Dis. 41, 499–513 (2018).
- Van Scherpenzeel, M., Steenbergen, G., Morava, E., Wevers, R. A. & Lefeber, D. J. High-resolution mass spectrometry glycoprofiling of intact transferrin for diagnosis and subtype identification in the congenital disorders of glycosylation. Transl. Res. 166, 639--649.e1 (2015).
- Sturiale, L., Barone, R. & Garozzo, D. The impact of mass spectrometry in the diagnosis of congenital disorders of glycosylation. J. Inherit. Metab. Dis. 34, 891–899 (2011).
- Bruneel, A., Cholet, S., Tran, N. T., Mai, T. D. & Fenaille, F. CDG biochemical screening: Where do we stand? Biochim. Biophys. Acta - Gen. Subj. 1864, (2020).
- Raynor, A. et al. Normal transferrin patterns in congenital disorders of glycosylation with Golgi homeostasis disruption: apolipoprotein C-III at the rescue! Clin. Chim. Acta 519, (2021).
- Taskinen, M. R. & Borén, J. Why Is Apolipoprotein CIII Emerging as a Novel Therapeutic Target to Reduce the Burden of Cardiovascular Disease? Current Atherosclerosis Reports vol. 18 (2016).
- Yassine, H. N. et al. The association of human apolipoprotein C-III sialylation proteoforms with plasma triglycerides. PLoS One 10, (2015).
- Francisco, R. et al. The challenge of CDG diagnosis. Molecular Genetics and Metabolism vol. 126 (2019).
- Fogli, A. et al. CSF N-Glycan Profiles to Investigate Biomarkers in Brain Developmental Disorders: Application to Leukodystrophies Related to eIF2B Mutations. PLoS One 7, e42688 (2012).
- Denecke, J. et al. Congenital disorder of glycosylation type Id: Clinical phenotype, molecular analysis, prenatal diagnosis, and glycosylation of fetal proteins. Pediatr. Res. 58, (2005).
- Jones, M. A. et al. Targeted polymerase chain reaction-based enrichment and next generation sequencing for diagnostic testing of congenital disorders of glycosylation. Genet. Med. 13, (2011).
- Schollen, E. et al. Clinical and molecular features of three patients with congenital disorders of glycosylation type Ih (CDG-Ih) (ALG8 deficiency). J. Med. Genet. 41, (2004).
- Jones, M. A. et al. Molecular diagnostic testing for congenital disorders of glycosylation (CDG): Detection rate for single gene testing and next generation sequencing panel testing. Mol. Genet. Metab. 110, (2013).
- Bewicke-Copley, F., Arjun Kumar, E., Palladino, G., Korfi, K. & Wang, J. Applications and analysis of targeted genomic sequencing in cancer studies. Computational and Structural Biotechnology Journal vol. 17 (2019).
- Park, J. H. & Marquardt, T. Treatment Options in Congenital Disorders of Glycosylation. Front. Genet. 12, 1716 (2021).
- Sosicka, P., Ng, B. G. & Freeze, H. H. Therapeutic Monosaccharides: Looking Back, Moving Forward. Biochemistry vol. 59 (2020).
- Brasil, S. et al. CDG therapies: From bench to bedside. International Journal of Molecular Sciences vol. 19 (2018).
- Janssen, M. C. H. et al. Successful liver transplantation and long-term follow-up in a patient with MPI-CDG. Pediatrics 134, (2014).
- Jansen, E. J. R. et al. ATP6AP1 deficiency causes an immunodeficiency with hepatopathy, cognitive impairment and abnormal protein glycosylation. Nat. Commun. 7, (2016).
- Jansen, J. C. et al. CCDC115 Deficiency Causes a Disorder of Golgi Homeostasis with Abnormal Protein Glycosylation. Am. J. Hum. Genet. 98, (2016).
- Jansen, J. C. et al. TMEM199 Deficiency is a Disorder of Golgi Homeostasis Characterized by Elevated Aminotransferases, Alkaline Phosphatase, and Cholesterol and Abnormal Glycosylation. Am. J. Hum. Genet. 98, (2016).
- Martínez-Monseny, A. F. et al. AZATAX: Acetazolamide safety and efficacy in cerebellar syndrome in PMM2 congenital disorder of glycosylation (PMM2-CDG). Ann. Neurol. 85, (2019).
- Yuste-Checa, P. et al. Pharmacological Chaperoning: A Potential Treatment for PMM2-CDG. Hum. Mutat. 38, (2017).
- Glycomine Programs PMM2-CDG. https://glycomine.com/programs/PMM2-CDG/.
- Pipeline | World CDG Organization. https://worldcdg.org/drug-development/pipeline.