
Lay Summary
The attachment of sugars (glycans) to proteins is known as protein glycosylation. Protein glycosylation can be further divided into two main categories, N-linked and O-linked protein glycosylation, depending on how sugars are added to proteins. O-linked protein glycosylation (or O-glycosylation) is the process by which glycans are added to specific amino acids, such as serine and threonine, in proteins. When a glycan is attached to a protein in this way, the product is called an O-linked glycoprotein. O-glycosylation requires a variety of different enzymes to activate, modify and attach sugars to proteins and can be classified by the first sugar that is attached to the amino acid in the protein. Any mutation that impairs O-glycosylation of proteins is known as a disorder of O-linked protein glycosylation. To date, almost 50 different types of congenital disorders of glycosylation (CDG) are caused by defects in the O-glycosylation pathway. Disorders of O-glycosylation present varied clinical features. Initial diagnostic testing involves screening of proteins for abnormal glycosylation patterns and analysis of muscle cells, but definitive diagnosis typically relies on genetic testing.
O-Linked Protein Glycosylation
O-linked protein glycosylation (or O-glycosylation) is a process where sugars are attached to the oxygen atom of certain amino acid residues in proteins – mainly serine and threonine, as well as others, such as tyrosine and hydroxylysine1–3. O-glycosylation happens once a protein has been synthesized and takes place primarily in the endoplasmic reticulum (ER) and Golgi Apparatus (Golgi); in some cases, it can also occur in the nucleus, cytoplasm, or mitochondria4,5. O-glycosylation is initiated by seven different monosaccharides that can be further extended to form longer sugar chains called O-glycans6. O-glycans are needed for protein folding and stability, as well as biological processes, such as cell signalling, cell attachment, and cell recognition7,8. Mutations in the genes involved in O-glycosylation can lead to CDG, which are classified as disorders of O-linked protein glycosylation.
Types of O-Glycosylation
There are seven different types of O-glycosylation which are classified based on initial monosaccharide that is attached to amino acid residue in the protein (Figure 1). These O-glycosylation types (and corresponding sugar) include9:
- O-mannosylation (mannose)
- O-xylosylation (xylose)
- O-fucosylation (fucose)
- O-GalNAcylation (N-acetylgalactosamine)
- O-GlcNAcylation (N-acetylglucosamine)
- O-glucosylation (glucose)
- O-galactosylation (galactose)
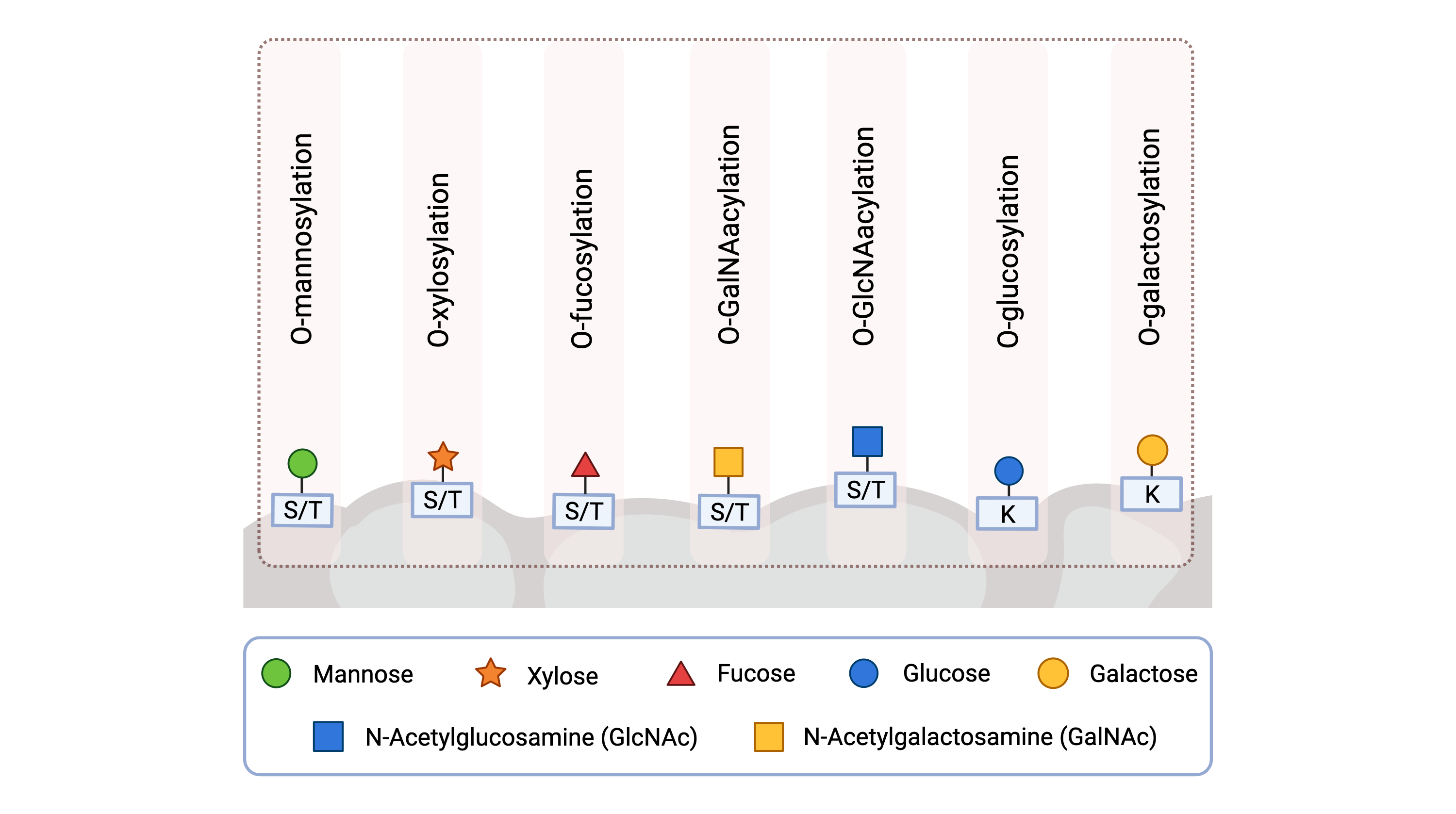
Figure 1: Different types of O-glycosylation in humans.
O-glycosylation can be classified by the first monosaccharide attached to the protein: O-mannosylation (mannose), O-xylosylation (xylose), O-fucosylation (fucose), O-GalNAcylation (GalNAc), O-GlcNAcylation (GlcNAc), O-glucosylation (glucose) and O-galactosylation (galactose). During O-glycosylation, a monosaccharide is attached onto specific amino acids of a protein, either serine (S), threonine (T) and lysine (K).
O-Glycosylation Pathways
Most O-glycosylation pathways can be divided into the following steps:
- Generation and transportation of activated sugars
- Monosaccharide attachment and O-glycan elongation
Generation and Transportation of Activated Sugars
Before O-glycosylation can occur, cells must generate high-energy (reactive) forms of monosaccharides, called activated sugars7. Activated sugars are also called glycosyl donors or sugar donors because they enable sugars to be added onto other molecules (Figure 2). Monosaccharides are activated by forming high-energy bonds to nucleosides (forming nucleotide sugars) or to dolichol phosphate (a lipid embedded in the ER membrane)10.
O-glycosylation can occur in different locations in the cell, such as the ER and Golgi, which are compartments surrounded by membranes that nucleotide sugars cannot pass through. Consequently, the ER and Golgi contain membrane proteins called nucleotide sugar transporters that shuttle nucleotide sugars from the cytosol to the inside of the ER or Golgi11,12.
Dolichol phosphate-linked monosaccharides are transported to the inside of the ER by a different method. Dolichol phosphate is embedded in the ER membrane and exposed to the cytosol. Consequently, a monosaccharide is transferred from a nucleotide sugar to dolichol phosphate on the cytosol side of the ER. The dolichol phosphate-linked monosaccharide is then flipped to the inside of the ER and the monosaccharide is transferred to the protein or glycan9,13.
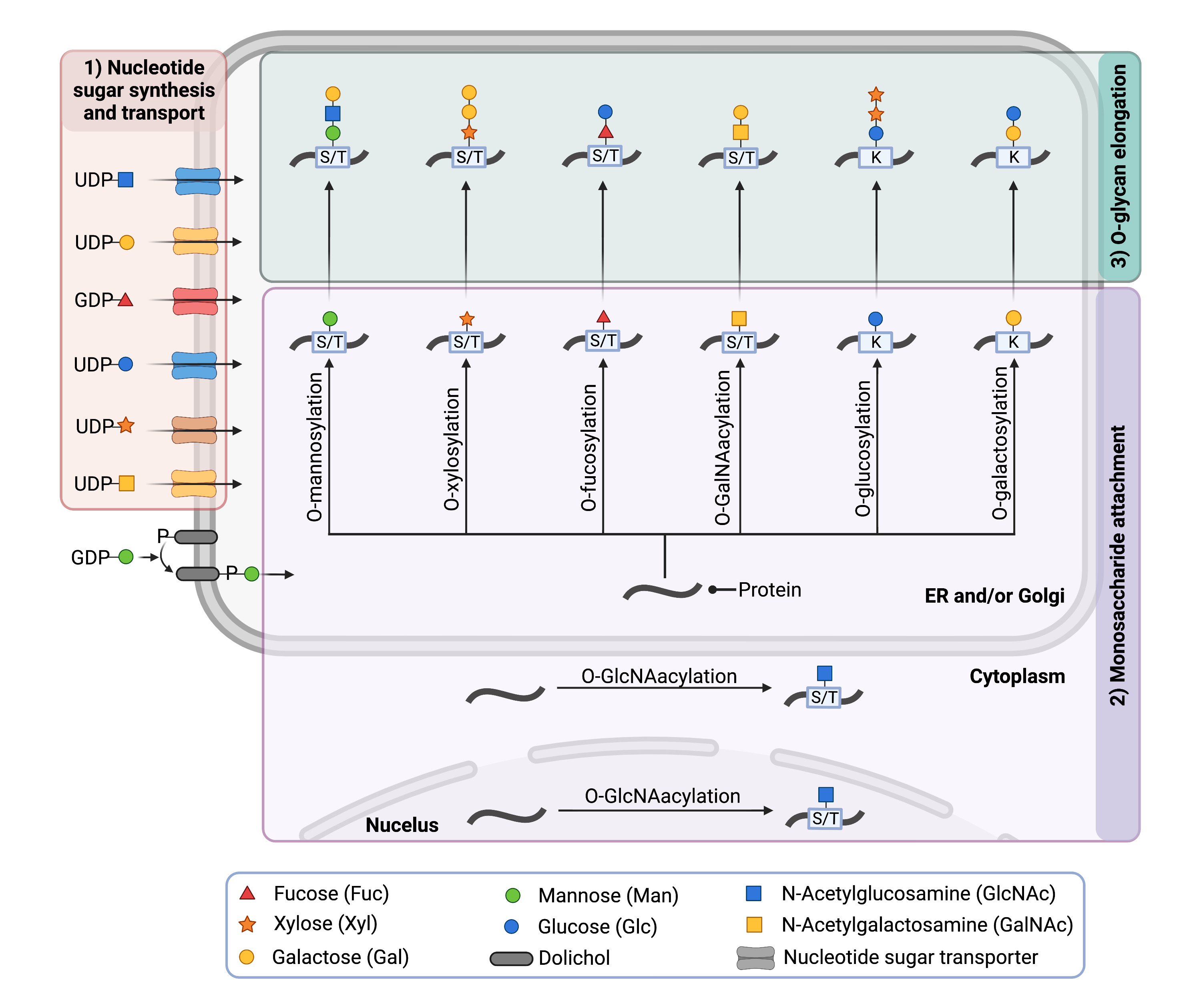
Figure 2. Overview of the O-glycosylation pathway.
During O-glycosylation, enzymes transfer monosaccharides from the activated sugars to amino acids (serine, threonine or lysine) on proteins, typically in the cytosol, Golgi, ER or nucleus. Enzymes may elongate the O-glycan chain in the Golgi by transferring additional monosaccharides from nucleotide sugars to the O-glycan chain.
Monosaccharide Attachment and O-Glycan Elongation
Enzymes called glycosyltransferases transfer the monosaccharide from the activated sugar to serine and threonine (or, to a lesser extent, tyrosine or hydroxylysine) residues on newly synthesized proteins—this process is called O-glycosylation9. O-glycans can be further elongated in the Golgi to generate complex O-glycan structures. As proteins travel through the compartments of the Golgi, additional monosaccharides are added to the growing glycan chain by glycosyltransferases12. Other modifications may involve adding sulfates (sulfotransferases), adding or removing phosphate groups (kinases or phosphatases, respectively), or altering the configuration of the monosaccharide (epimerases)14–17. Synthases are used in O-glycosylation to generate substrates, such as monosaccharide-based substrates18.
Disorders of O-Glycosylation
Disorders of O-glycosylation arise from abnormalities in O-glycosylation pathways, and 47 are currently known. Most O-linked CDG are inherited in an autosomal recessive fashion, with affected individuals receiving one defective allele from each asymptomatic parent19.
Disorders of O-glycosylation can be grouped based on the type of O-glycan that is affected (Figure 3). For example, if a deficient enzyme prevents the formation of a normal glycan structure that is attached to a protein by a mannose sugar, then it is categorized as a disorder of O-mannosylation. Each class of O-glycosylation relies on variety of different types of enzymes, and disorders of O-glycosylation are caused by mutations in the genes encoding these enzymes20.
O-linked CDG that have been identified to date include:
- Disorders of O-Mannosylation
- Disorders of O-Xylosylation (GAG biosynthesis)
- Disorders of O-Fucosylation
- Disorders of O-GalNAcylation
- Disorders of O-GlcNAcylation
- Disorders of O-Glucosylation
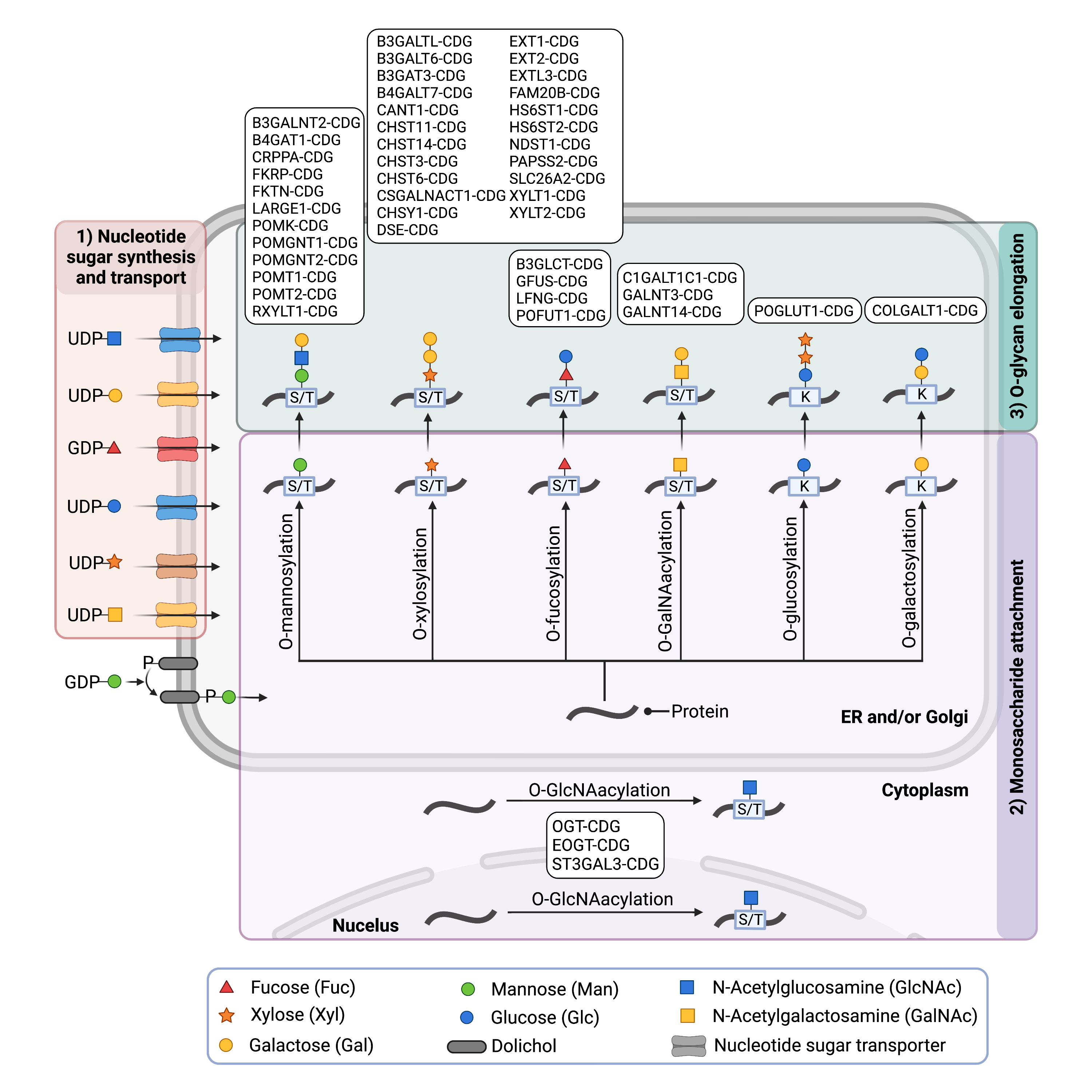
Figure 3. Overview of CDG involving O-glycosylation Defects.
O-linked CDG may be classified into groups based on the type of O-glycan that is affected.
Disorders of O-Mannosylation
Disorders of O-mannosylation are caused by mutations in genes that encode proteins involved in the attachment of mannose onto proteins and the elongation of glycan chains on O-mannosylated glycoproteins7. These disorders particularly impact alpha-dystroglycan, a heavily O-mannosylated glycoprotein. Cells use alpha-dystroglycan to interact with their surroundings, such as cell attachment and cellular signalling, and it is particularly abundant in skeletal muscle and the brain21. Alpha-dystroglycan must be sufficiently glycosylated for attachment and signalling22. Thus, disorders of O-mannosylation are typically associated with dystroglycanopathies, the term given to the group of diseases that develop from insufficient glycosylation levels of alpha-dystroglycan23. These disorders have a unique presentation that affects the muscles and may range from severe muscular dystrophy to milder muscle abnormalities. As with many CDG, the clinical presentations may include brain and eye abnormalities21.
Disorders of O-mannosylation include the following:
Disorders of O-Xylosylation (GAG Biosynthesis)
Disorders of O-xylosylation are caused by mutations in genes that encode proteins involved in the attachment of xylose onto proteins and the building up of the glycan chains on O-xylosylated glycoproteins, as well as nucleotide sugar transporters that ensure adequate supply of sugars needed for O-xylosylation. These defects can impact the biosynthesis of glycosaminoglycans (also referred to as GAGs) which are long glycan chains with repeating glycan structures that are often attached to proteins, forming proteoglycans. Some types of glycosaminoglycans have a xylose residue at the end of their glycan chains, which is attached to the protein when generating the corresponding proteoglycan24. Proteoglycans are used in a variety of biological processes, including cell signalling, cell attachment and development24,25.
Abnormalities in O-xylosylation (GAG biosynthesis) can cause disorders, such as hereditary multiple exostoses and Ehlers–Danlos syndrome26,27, and include:
|
Disorders of O-Fucosylation
Disorders of O-fucosylation are caused by mutations in genes that provide the code for proteins involved in the attachment of fucose onto proteins and the building up of the glycan chains on O-fucosylated glycoproteins28. Disorders of O-fucosylation can be associated with different disorders, such as Peter Plus syndrome and Dowling Degos disease28,29. Disorders of O-fucosylation include:
Disorders of O-Galnacylation
O-GalNAc glycans are one of the most common types of O-glycans and are highly abundant on a class of glycoproteins called mucins. O-GalNAc glycans, also called mucin-type O-glycans, can be extended to form one of four core structures.
Disorders of O-GalNAcylation are caused by mutations in genes that encode for proteins involved in the attachment of N-acetylgalactosamine (GalNAc) onto proteins and the building up of the glycan chains on O-GalNAcylated glycoproteins. CDG caused by defects in O-GalNAcylation include30–32:
Disorders of O-GlcNAcylation
Disorders of O-GlcNAcylation are caused by mutations in genes that provide the code for proteins involved in the attachment of N-acetylglucosamine (GlcNAc) onto proteins and the building up of the glycan chains on O-GlcNAcylated glycoproteins, and they are associated with Adams-Oliver syndrome33,34. Disorders of O-GlcNAcylation include:
Disorders of O-Glucosylation
The only currently known disorder of O-glucosylation arises from mutations in POGLUT1, a gene encoding a glucosyltransferase that transfers glucose onto serine residues of some proteins, which can cause muscular dystrophy and Dowling-Degos disease 432,35. The corresponding CDG is known as:
Disorders of O-Galactosylation
The only currently known disorder of O-galactosylation arises from mutations in COLGALT1, a gene encoding a galactosyltransferase that transfers galactose onto a hydroxyline residues of collagen. Mutations in COLGALT1 cause brain small vessel disease 3, a disorder resulting from fragile cerebral vessels36. The corresponding CDG is known as:
Diagnosis
Like all CDG, disorders of O-linked glycosylation vary widely in their clinical presentation but typically manifest symptoms in multiple systems. Common presentations include neurological abnormalities and developmental/cognitive delay, failure to thrive, poor muscle tone (hypotonia), muscular dystrophy, and clotting abnormalities. Cardiac, ophthalmological, dermatological, and renal abnormalities may also be present37.
Diagnosis of O-linked CDG is achieved by conducting a series of tests which can help narrow down which part of the glycosylation pathway is affected and potentially identify individual CDG. O-linked glycosylation disorders cannot be detected with transferrin analysis, as the transferrin protein is only modified with N-glycans (glycans linked to proteins through a nitrogen atom on asparagine residues). However, patients may be assessed for abnormalities with O-glycosylated proteins, such as apolipoprotein C-III, or by analysis of analysis of total O-glycan content present in plasma or serum38. O-glycan defects linked to alpha-dystroglycanopathies (disorders of O-mannosylation), can be identified by specialized staining techniques that use antibodies to bind to glycosylated alpha-dystroglycan39. Disorders affecting GAG biosynthesis (disorders of O-xylosylation) can also be identified using experiments that use antibodies to detect defects in O-linked proteoglycans40. However, diagnosis of suspected individuals with O-linked glycosylation disorders typically relies on genetic testing strategies41.
Apolipoprotein C-III Analysis
Apolipoprotein C-III (apo C-III) is a glycoprotein found in the blood that has a single O-linked glycan (a core 1 mucin-type O-glycan) attached to a threonine residue (Figure 4) 42. Analysis of apolipoprotein C-III can reveal disorders involving O-GalNacylation 43. ApoC-III can exist in three isoforms (glycofoms) with different numbers of sialic acid residues, including zero (apoC-III0), one (apoC-III1) or two (apoC-III2) sialic acid residues44.
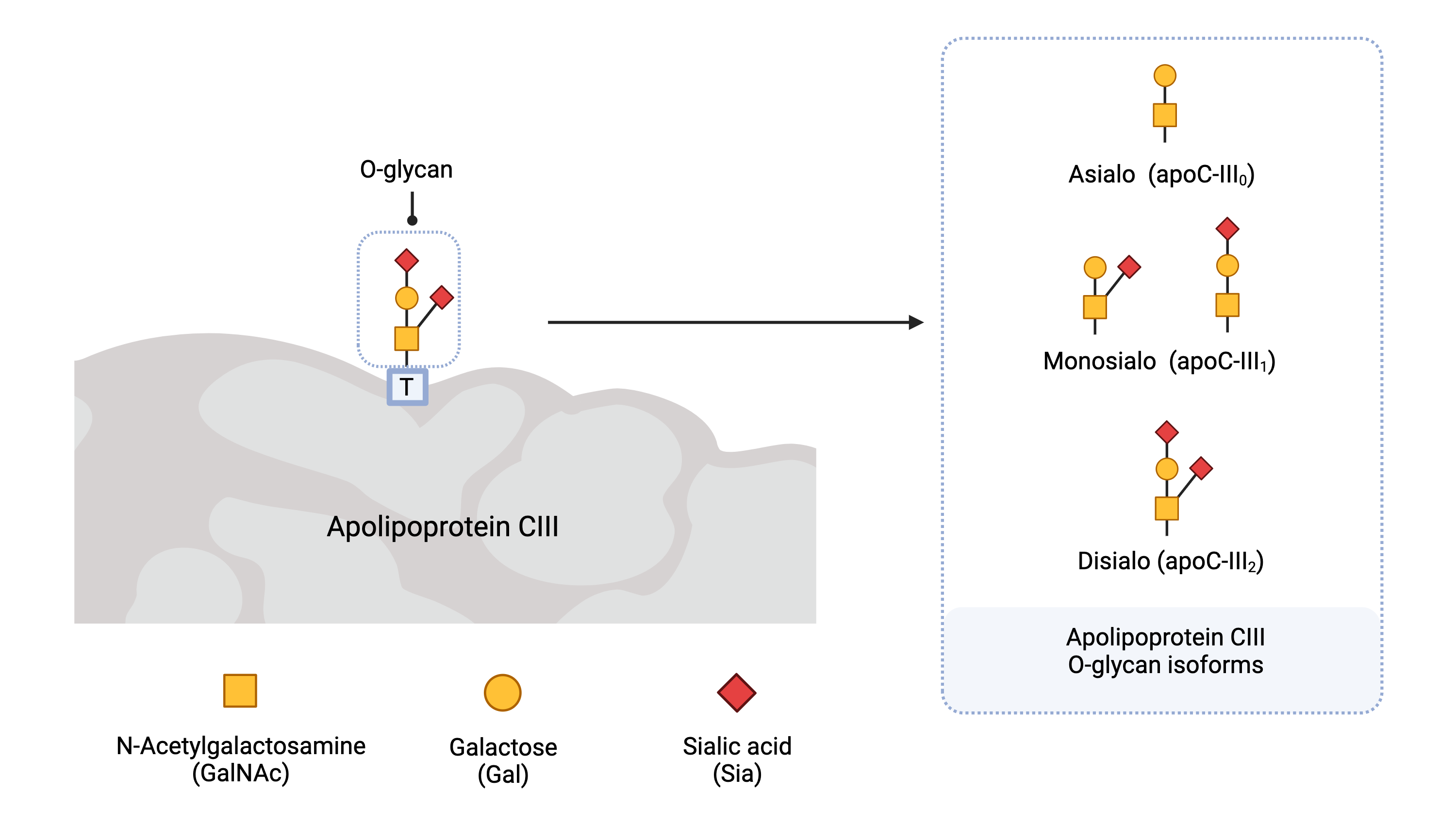
Figure 4. Apolipoprotein C-III isoforms.
Apolipoprotein C-III (Apo C-III) contains a single O-glycan and is present in different isoforms (glycoforms) depending the number of sialic acid residues present on the glycan. Apo C-III isoforms are named according to the number of sialic acid residues present.
CDG that arise from defects in O-GalNAcylation or defects in Golgi homeostasis have increased levels of apo C-III0 and/or apo C-III145. These can be detected by different methods, such as isoelectric focusing (IEF) (Figure 5), two-dimensional gel electrophoresis, capillary electrophoresis and mass spectrometry45,46.
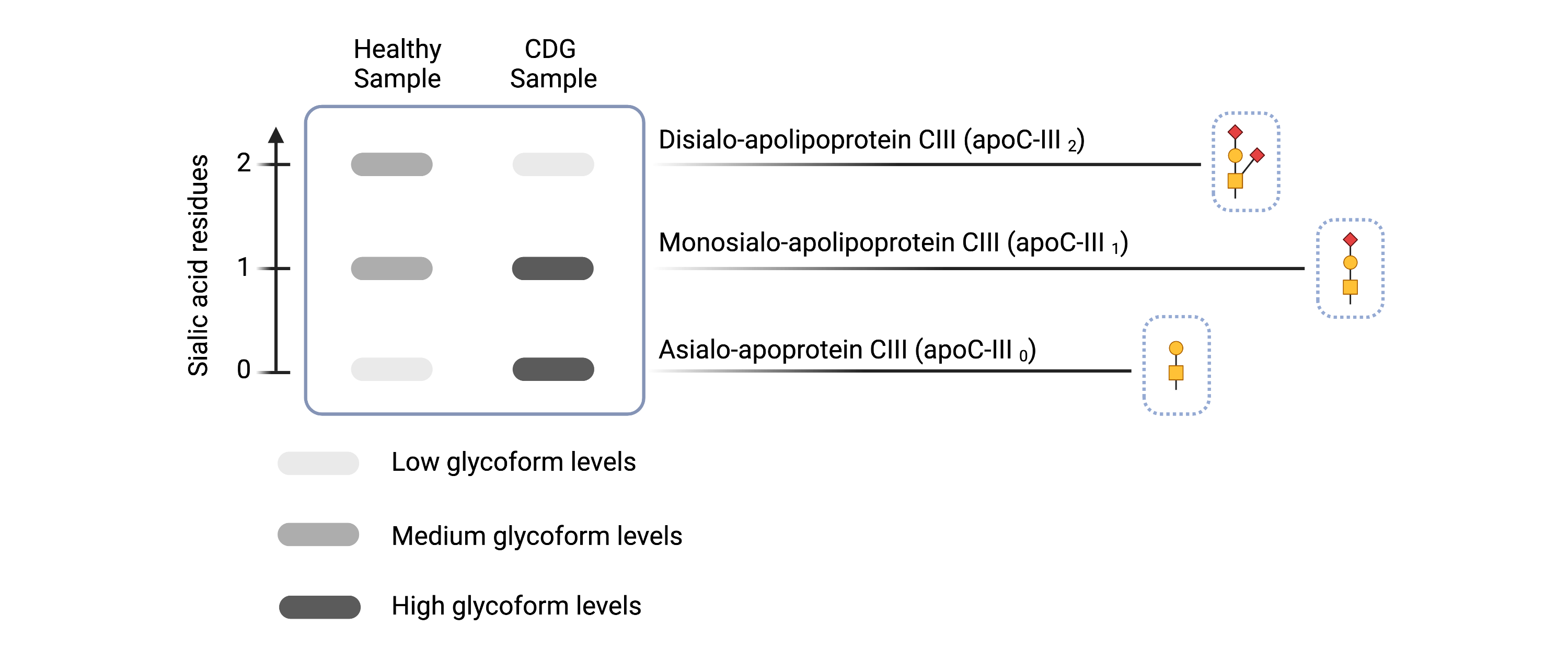
Figure 5. Example of Apolipoprotein C-III isoelectric focusing results for healthy and CDG samples.
Apolipoprotein C-III isoelectric focusing can detect some O-linked CDGs and CDGs that involve Golgi defects by the presence of abnormal isoform levels in patient serum samples.
Total O-Glycan Analysis
Total O-glycan analysis can be assessed using mass spectrometry to detect abnormal O-glycans, typically from blood samples47.
Genetic Testing
Genetic testing is often required to definitively diagnose O-linked CDG and may include targeted sequencing of specific genes, use of CDG gene panels that test for known O-linked CDG, whole genome sequencing (WGS), and the sequencing of all expressed genes (whole exome sequencing, WES)47–51.
References
- Reily, C., Stewart, T. J., Renfrow, M. B. & Novak, J. Glycosylation in health and disease. Nature Reviews Nephrology 15, (2019).
- Halim, A. et al. Site-specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid -peptides in human cerebrospinal fluid. Proceedings of the National Academy of Sciences 108, (2011).
- Tang, M. et al. Effect of hydroxylysine-O-glycosylation on the structure of type I collagen molecule: A computational study. Glycobiology 30, (2020).
- Li, F. et al. GlycoMinestruct: a new bioinformatics tool for highly accurate mapping of the human N-linked and O-linked glycoproteomes by incorporating structural features. Scientific Reports 6, 34595 (2016).
- Yang, X. & Qian, K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nature Reviews Molecular Cell Biology 18, (2017).
- Stanley, P. Golgi glycosylation. Cold Spring Harbor Perspectives in Biology 3, (2011).
- van Tol, W., Wessels, H. & Lefeber, D. J. O-glycosylation disorders pave the road for understanding the complex human O-glycosylation machinery. Current Opinion in Structural Biology 56, 107–118 (2019).
- Saentaweesuk, W. et al. Clinical significance of GalNAcylated glycans in cholangiocarcinoma: values for diagnosis and prognosis. Clinica Chimica Acta 477, (2018).
- Wopereis, S., Lefeber, D. J., Morava, É. & Wevers, R. A. Mechanisms in Protein O-Glycan Biosynthesis and Clinical and Molecular Aspects of Protein O-Glycan Biosynthesis Defects: A Review. Clinical Chemistry 52, (2006).
- Grünewald, S., Matthijs, G. & Jaeken, J. Congenital disorders of glycosylation: A review. Pediatric Research vol. 52 (2002).
- Hadley, B. et al. Structure and function of nucleotide sugar transporters: Current progress. Computational and Structural Biotechnology Journal 10, (2014).
- Reily, C., Stewart, T. J., Renfrow, M. B. & Novak, J. Glycosylation in health and disease. Nature Reviews Nephrology 15, (2019).
- Neubert, P. & Strahl, S. Protein O-mannosylation in the early secretory pathway. Current Opinion in Cell Biology 41, (2016).
- Martínez Cuesta, S., Rahman, S. A., Furnham, N. & Thornton, J. M. The Classification and Evolution of Enzyme Function. Biophysical Journal 109, (2015).
- Schjoldager, K. T., Narimatsu, Y., Joshi, H. J. & Clausen, H. Global view of human protein glycosylation pathways and functions. Nature Reviews Molecular Cell Biology 21, (2020).
- Walimbe, A. S. et al. Pomk regulates dystroglycan function via large1-mediated elongation of matriglycan. eLife 9, (2020).
- Dubail, J. & Cormier-Daire, V. Chondrodysplasias With Multiple Dislocations Caused by Defects in Glycosaminoglycan Synthesis. Frontiers in Genetics vol. 12 (2021).
- Riemersma, M. et al. Human ISPD Is a Cytidyltransferase Required for Dystroglycan O-Mannosylation. Chemistry & Biology 22, (2015).
- Chang, I. J., He, M. & Lam, C. T. Congenital disorders of glycosylation. Annals of Translational Medicine 6, (2018).
- Jaeken, J. & Péanne, R. Erratum to: What is new in CDG? Journal of Inherited Metabolic Disease 40, (2017).
- Stevens, E. et al. Mutations in B3GALNT2 Cause Congenital Muscular Dystrophy and Hypoglycosylation of α-Dystroglycan. The American Journal of Human Genetics 92, 354–365 (2013).
- Brancaccio, A. A molecular overview of the primary dystroglycanopathies. Journal of Cellular and Molecular Medicine 23, (2019).
- Endo, T. Glycobiology of alpha-dystroglycan and muscular dystrophy. Journal of Biochemistry 157, (2015).
- Müller, T. et al. Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers–Danlos syndrome. Human Molecular Genetics 22, (2013).
- Paganini, C., Costantini, R., Superti‐Furga, A. & Rossi, A. Bone and connective tissue disorders caused by defects in glycosaminoglycan biosynthesis: a panoramic view. The FEBS Journal 286, (2019).
- Brady, A. F. et al. The Ehlers-Danlos syndromes, rare types. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 175, (2017).
- Al-Zayed, Z. et al. Mutation spectrum of EXT1 and EXT2 in the Saudi patients with hereditary multiple exostoses. Orphanet Journal of Rare Diseases 16, (2021).
- Wang, Y. E., Ramirez, D. A., Chang, T. C. & Berrocal, A. Peters plus syndrome and Chorioretinal findings associated with B3GLCT gene mutation - a case report. BMC Ophthalmology 20, (2020).
- Chen, M. et al. Analysis of POFUT1 Gene Mutation in a Chinese Family with Dowling-Degos Disease. PLoS ONE 9, (2014).
- Zilmer, M. et al. Novel congenital disorder of O -linked glycosylation caused by GALNT2 loss of function. Brain 143, (2020).
- Freeze, H. H., Schachter, H. & Kinoshita, T. Genetic disorders of glycosylation. in Essentials of Glycobiology (eds. Varki, A., Cummings, R. & Esko, J.) ( Cold Spring Harbor Laboratory Press, 2017).
- Ferreira, C. R. et al. Recognizable phenotypes in CDG. Journal of Inherited Metabolic Disease vol. 41 (2018).
- Jaeken, J. Congenital disorders of glycosylation: A multi-genetic disease family with multiple subcellular locations. Journal of mother and child 24, (2020).
- Hu, H. et al. ST3GAL3 mutations impair the development of higher cognitive functions. American Journal of Human Genetics 89, (2011).
- Servián‐Morilla, E. et al. A POGLUT1 mutation causes a muscular dystrophy with reduced Notch signaling and satellite cell loss. EMBO Molecular Medicine 8, (2016).
- Miyatake, S. et al. Biallelic COLGALT1 variants are associated with cerebral small vessel disease. Annals of Neurology 84, (2018).
- Péanne, R. et al. Congenital disorders of glycosylation (CDG): Quo vadis? European Journal of Medical Genetics 61, (2018).
- Ganetzky, R., Reynoso, F. J. & He, M. Congenital disorders of glycosylation. in Biomarkers in Inborn Errors of Metabolism (Elsevier, 2017).
- Johnson, K. et al. Detection of variants in dystroglycanopathy-associated genes through the application of targeted whole-exome sequencing analysis to a large cohort of patients with unexplained limb-girdle muscle weakness. Skeletal Muscle 8, (2018).
- Amadi, B. et al. Reduced production of sulfated glycosaminoglycans occurs in Zambian children with kwashiorkor but not marasmus. American Journal of Clinical Nutrition 89, (2009).
- Chang, I. J., He, M. & Lam, C. T. Congenital disorders of glycosylation. Annals of Translational Medicine 6, (2018).
- Taskinen, M. R. & Borén, J. Why Is Apolipoprotein CIII Emerging as a Novel Therapeutic Target to Reduce the Burden of Cardiovascular Disease? Current Atherosclerosis Reports vol. 18 (2016).
- Heywood, W. E. et al. Global serum glycoform profiling for the investigation of dystroglycanopathies & Congenital Disorders of Glycosylation. Molecular Genetics and Metabolism Reports 7, (2016).
- Yassine, H. N. et al. The association of human apolipoprotein C-III sialylation proteoforms with plasma triglycerides. PLoS ONE 10, (2015).
- Raynor, A. et al. Normal transferrin patterns in congenital disorders of glycosylation with Golgi homeostasis disruption: apolipoprotein C-III at the rescue! Clinica Chimica Acta 519, (2021).
- Francisco, R. et al. The challenge of CDG diagnosis. Molecular Genetics and Metabolism vol. 126 (2019).
- Abu Bakar, N., Lefeber, D. J. & van Scherpenzeel, M. Clinical glycomics for the diagnosis of congenital disorders of glycosylation. Journal of Inherited Metabolic Disease 41, (2018).
- Francisco, R. et al. The challenge of CDG diagnosis. Molecular Genetics and Metabolism 126, 1–5 (2019).
- Timal, S. et al. Gene identification in the congenital disorders of glycosylation type i by whole-exome sequencing. Human Molecular Genetics 21, 4151–4161 (2012).
- Bruneel, A., Cholet, S., Tran, N. T., Mai, T. D. & Fenaille, F. CDG biochemical screening: Where do we stand? Biochimica et Biophysica Acta - General Subjects 1864, (2020).
- Chang, I. J., He, M. & Lam, C. T. Congenital disorders of glycosylation. Annals of Translational Medicine 6, 1–13 (2018).