
Lay Summary
ALG1-CDG, formerly known as CDG-Ik, is a rare inherited condition that affects many parts of the body. Almost 60 cases of ALG1-CDG have been reported in the literature to date. ALG1-CDG is classified as a disorder of N-linked protein glycosylation. ALG1-CDG is caused when an individual has mutations in both copies of the ALG1 gene, which provides instructions for making an enzyme that attaches the simple sugar mannose to growing sugar chains during N-glycosylation. Mutations in the ALG1 gene cause proteins to be under glycosylated. Symptoms of ALG1-CDG begin at infancy and are primarily characterized by neurological abnormalities including developmental delay and low muscle tone, abnormal features, gastrointestinal and ophthalmological problems. Several screening tests are available for ALG1-CDG, and a biomarker has been identified, but a definitive diagnosis is achieved through genetic testing. Treatment is focused on the management of specific symptoms and preventing complications.
Overview
Beta-1,4-mannosyltransferase congenital disorder of glycosylation (ALG1-CDG) is a rare autosomal recessive genetic disorder1. The first reported case of ALG1-CDG was in 20041–3 and almost 60 cases have been reported cases in the literature4–10. The ALG1 (asparagine-linked glycosylation 1) gene encodes an enzyme responsible for adding the first of nine mannose residues during lipid linked oligosaccharide (LLO) synthesis on the cytoplasmic side of the endoplasmic reticulum (ER)5,6. LLO synthesis is a precursor step to N-glycosylation. Deficiency in the ALG1 enzyme results in the incomplete assembly of the LLO, leading to insufficient N-glycosylation of glycoproteins.
Symptoms begin at infancy and the characteristic presentations of ALG1-CDG include neurological symptoms, abnormal facial features, gastrointestinal and ophthalmological problems5,6. A diagnosis can be determined through transferrin analysis, LLO analysis, and detection of xeno-tetrasaccharide – a unique oligosaccharide found on serum glycoproteins and transferrin in individuals with ALG1-CDG which may serve as a biomarker for this CDG5,11. Definitive diagnosis can only be achieved through molecular genetic testing. There are currently no approved treatments for ALG1-CDG6.
Synonyms
- CDG-Ik
- Asparagine-linked glycosylation 1
- HMT1, human mannosyltransferase 1
Inheritance
ALG1-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the genefrom each asymptomatic parent5.
Gene Function
ALG1 encodes a mannosyltransferase enzyme, beta-1,4-mannosyltransferase (ALG1)5. Mannosyltransferases are enzymes that enable the transfer of mannose during glycosylation. ALG1 is located in the ER membrane, facing the cytosol, where it has a role in the assembly of the LLO, a precursor for protein N-glycosylation6. The ALG1 enzyme transfers the first of nine mannose residues to the growing LLO prior to its attachment to a protein. Mutations in ALG1 cause reduced enzymatic activity1.
LLO synthesis
N-glycosylation is the process by which an oligosaccharide is attached to the nitrogen atom of asparagine residues on proteins. N-glycosylation is initiated in the ER and begins with the synthesis of an LLO12. The LLO is comprised of a 14-sugar oligosaccharide attached to the lipid carrier dolichol pyrophosphate (Dol-PP). The 14-sugar glycan chain is made up of 2 N-acetylglucosamine (GlcNAc), 9 mannose, and 3 glucose residues. Once assembled, the oligosaccharide is transferred "en bloc" to proteins and undergoes further processing in the ER and Golgi12. Once the oligosaccharide is attached to a protein, it is referred to as an N-glycan.
LLO synthesis is carried out by a series of enzymes encoded mostly by the ALG genes and can be divided into two phases: Phase I and Phase II 12 (Figure 1).
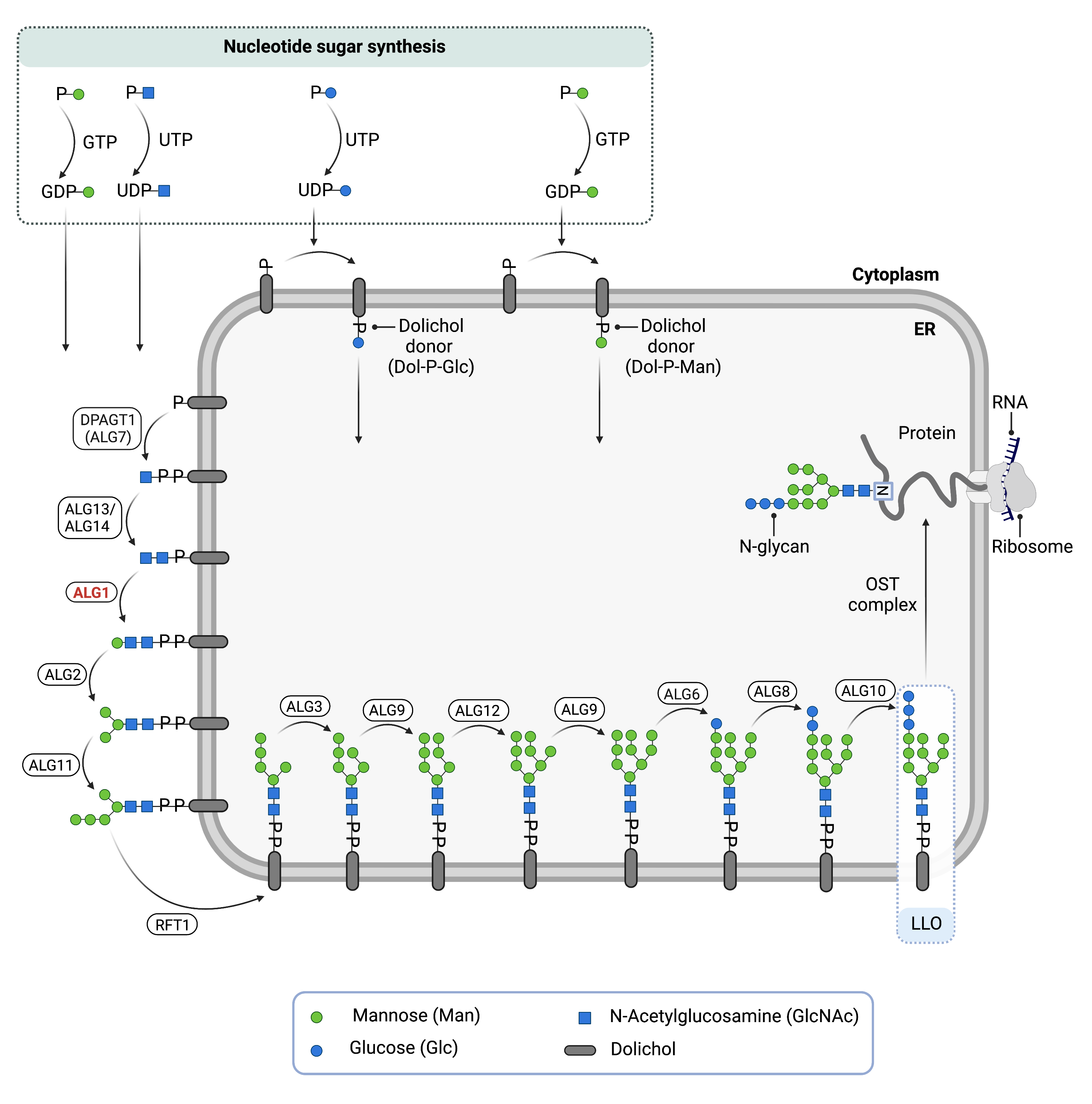
Figure 1. Role of ALG1 in glycosylation.
ALG1 is an enzyme (beta-1,4-mannosyltransferase) involved in synthesizing the lipid-linked oligosaccharide (LLO) in N-glycosylation. ALG1 adds mannose to the LLO on the cytosolic side of the endoplasmic reticulum membrane.
Phase I
Phase I of LLO synthesis takes place on the cytoplasmic side of the ER. It begins with the sequential attachment of two N-acetylglucosamine (GlcNAc2) and five mannose (Man5) residues to Dol-PP by several ER enzymes. GlcNAc and mannose are transferred from nucleotide sugars UDP-acetylglucosamine and GDP-mannose, respectively. The intermediate structure, Dol-PP-GlcNAc2Man5, is translocated from the cytoplasm into the ER lumen the RFT1 enzyme12,13.The ALG1 enzyme is responsible for adding the first mannose residue to the growing LLO14.
Phase II
Phase II of LLO synthesis takes place in the ER lumen. Once in the lumen, four mannose residues followed by three glucose (Glc3) residues are added to the intermediate structure, generating the complete N-glycan, Dol-PP-GlcNAc2Man9Glc3. Mannose and glucose are transferred from glycosyl donors Dol-P-mannose and Dol-P-glucose, which are formed on the cytoplasmic side of the ER and must also be flipped across the ER membrane12.
Once assembled, the oligosaccharide is transferred “en bloc” from Dol-PP to asparagine residues of newly synthesized protein via the enzyme oligosaccharyltransferase (OST), resulting in N-glycosylation of the protein12. The activity of OST is highly specific for the completely assembled 14-sugar oligosaccharide, Glc3Man9GlcNAc2.
Disease Mechanism
Mutations in the ALG1 gene lead to the production of an abnormal enzyme with reduced activity1. In the absence of a functional ALG1, the LLO is missing nine mannose and three glucose residues and is therefore incompletely assembled. This results in reduced transfer efficiency of the oligosaccharide by OST, causing an accumulation of the incomplete LLO, GlcNAc2-PP-dolichol5, and hypoglycosylation of glycoproteins which lack N-glycans13.
Mutations
The ALG1 gene is located on chromosome 16 (16p13.3). There are 43 pathogenic variants reported in the ALG1 gene15. The most common mutation is c. 733C>T (p.Ser258Leu)16 which has been suggested to be a possible founder mutation and is associated with a severe phenotype; whereas the second most common mutation (c. 1076C>T) may be associated with a milder phenotype9,16. All individuals homozygous for the p.Ser258Leu mutation died before 6 months of age5.
Signs & Symptoms
Clinical Presentation
Individuals with ALG1-CDG develop signs and symptoms during infancy, as early as a few days old. ALG1-CDG is primarily characterized by mild to severe neurological disorder and multi-system organ involvement17. The characteristic clinical presentations of ALG1-CDG include4–6,16,18:
- Neurological – psychomotor developmental delay, intellectual disability, low muscle tone (hypotonia), epilepsy, and a smaller than average head at birth (microcephaly)
- Dysmorphic features – small lower jaw, abnormally shaped and low-set ears, large forehead, small mouth and thin lips, small and almond shaped eyes, and a widened nasal bridge
- Gastrointestinal – intestinal problems may lead to failure to gain weight and slower than normal growth (failure to thrive). Protein-losing enteropathy (PLE) and chronic diarrhea are the most common gastrointestinal problems. PLE is one of the more life-threatening symptoms, causing an excess loss of proteins in the gastrointestinal tract
- Ophthalmological – misaligned or crossed eyes (strabismus), repetitive and uncontrolled eye movements (nystagmus), retinopathy, and vision loss
Less common clinical presentations of ALG1-CDG include abnormal brain imaging including cerebral or cerebellar atrophy, skeletal abnormalities including scoliosis, curving of the spine (kyphosis), and joint contractures, and abnormal fat distribution5.
Biochemical Abnormalities
Biochemical abnormalities observed in ALG1-CDG patients include hematological defects such as anemia and abnormal clotting, low levels of albumin in the blood (hypoalbuminemia), elevated liver enzymes, increased serum transaminases, decreased serum LDL cholesterol, cholinesterase, and immunoglobulins15. Hypoalbuminemia is a severe symptom of ALG1-CDG, as all patients reported with hypoalbuminemia as a symptom died at an average age of 6.75 months5.
Classification
ALG1-CDG is classified as a disorder of N-linked protein glycosylation.
Under the former CDG classification system, ALG1-CDG is classified as a Type 1 CDG, which arise due to defects in the synthesis of oligosaccharides or their transfer to proteins.
Diagnosis
Although diagnosis of ALG1-CDG may be suspected based on presentation of symptoms and detailed patient history, direct molecular genetic testing is the only definitive diagnostic test. Screening in suspected patients begins with a blood test to analyze serum transferrin. LLO analysis in patient fibroblasts may also be carried out following transferrin screening. Additionally, a protein-linked oligosaccharide biomarker, xeno-tetrasaccharide, has been observed in all ALG1-CDG patients who have been tested for this marker5.
Transferrin Analysis
Individuals with ALG1-CDG show a type 1 pattern by transferrin isoelectric focusing (TIEF) or mass spectrometry analysis of transferrin6. Type 1 patterns are observed in CDG that arise due to defects in the LLO synthesis and are characterized by an increase in disialo- and asialo-transferrin isoforms19.
LLO Analysis
LLO analysis of ALG1-CDG patient fibroblasts show an increase of the intermediate LLO structure Dol-PP-GlcNAc2, which lacks mannose residues15.
Biomarkers
NeuAc-Gal-GlcNAc2 (xeno-tetrasaccharide)
Serum, plasma, and patient-derived fibroblasts may be tested for the biomarker xeno-tetrasaccharide NeuAc-Gal-GlcNAc2: a four-sugar oligosaccharide comprised of two GlcNAc, one galactose (Gal), and one sialic acid (NeuAc). NeuAc-Gal-GlcNAc2 is a novel N-linked oligosaccharide present on glycoproteins, including transferrin, that accumulates in individuals with ALG1-CDG. All ALG1-CDG patients have shown a presence of this biomarker, which has only been detected in trace amounts in other CDG patients (PMM2-CDG and MP1-CDG)4,5,11,20.
Prognosis
Prognosis of ALG1-CDG may vary depending on the severity of an individual’s symptoms. Severe forms of ALG1-CDG can cause early death but there are also reports in the medical literature of patients surviving to adulthood. The ALG1-CDG mutation c.773C>T is associated with severe forms of ALG1-CDG2,6. Premature death was caused by respiratory or renal failure, or infections leading to sepsis5. Other patients with more mild symptoms show neurological disabilities, microcephaly, and seizures6.
Management
Management of symptoms may include combinations of physical therapy, occupational therapy, speech or vision therapy, and palliative measures6.
Therapies
There are currently no treatment options available for ALG1-CDG. Treatment is focused on management of symptoms and prevention of complications6.
Research Models
Several ALG1 research models have been generated including yeast, fly, and mouse models.
Yeast (S. cerevisiae)
Alg1 mutants (strains K57-6C and PRY56) have been used in complementation assays to observe the effects of mutation on ALG1 protein function3,5,6,21. Both strains are thermosensitive. The K57-6C strain shows increased levels of GDP-Man by mannose-1-phosphate as well as overexpression of a guanylyltransferase gene (Mpg1) which restores defects in mannosylation22.
Fly (D. melanogaster) Mouse
Drosophila gene Alg1 (ALG1, CG18012, FBgn0038552) is orthologous to ALG1. It is predicted to have mannosyltransferase activity and be involved in protein glycosylation. It is expressed in the adult drosophila head and is a potential model for human disease ALG1-CDG (FlyBase).
Mouse (M. musculus)
Alg1-/- knockout mouse
B6/JGpt-Alg1em1Cflox/Gpt is an available coisogenic mutant strain (IMSR). Alg1tm1b(KOMP)Wtsi mice have a reporter-tagged deletion allele in which mice either homozygous or heterozygous for the mutant allele live to early adulthood. Homozygous mice have a preweaning lethality phenotype with complete penetrance, while heterozygous mice have abnormal coat/hair pigmentation (IMPC).
ALG1 conditional-ready floxed knockout embryonic stem cell line
Embryonic stem (ES) cell line Alg1tm1a(KOMP)Wtsi is a targeted knockout/null mutation of Alg1. A “conditional-ready” allele can be created by flp recombinase expression in mice carrying this allele, and cre expression results in a knockout mouse. If cre is expressed without flp expression, a reporter knockout mouse can be generated (MGI).
ALG1 knockout ES cell line
ES cell line Alg1tm1e(KOMP)Wtsi is a targeted knockout/null mutation of Alg1 (MGI).
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including ALG1-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Organizations
ALG1-CDG Families Facebook Group
Publications
ALG1-CDG Scientific Articles on PubMed
Additional Resources
ALG1-CDG Clinical Utility Gene Card
OMIM
OrphaNet
NORD
GARD
MedlinePlus
Genetic Testing Registry
ClinVar
NIH
GeneCards
UniProt
References
- Schwarz, M. et al. Deficiency of GDP-Man:GlcNAc2-PP-Dolichol Mannosyltransferase Causes Congenital Disorder of Glycosylation Type Ik. The American Journal of Human Genetics 74, (2004).
- Kranz, C. et al. Congenital Disorder of Glycosylation Type Ik (CDG-Ik): A Defect of Mannosyltransferase I. The American Journal of Human Genetics 74, (2004).
- Grubenmann, C. E. Deficiency of the first mannosylation step in the N-glycosylation pathway causes congenital disorder of glycosylation type Ik. Human Molecular Genetics 13, (2004).
- González-Domínguez, C. A. et al. ALG1-CDG Caused by Non-functional Alternative Splicing Involving a Novel Pathogenic Complex Allele. Frontiers in Genetics 12, (2021).
- Ng, B. G. et al. ALG1-CDG: Clinical and Molecular Characterization of 39 Unreported Patients. Human Mutation37, (2016).
- Rohlfing, A.-K. et al. ALG1-CDG: A new case with early fatal outcome. Gene 534, (2014).
- Snow, T. M., Woods, C. W. & Woods, A. G. Congenital Disorder of Glycosylation. Advances in Neonatal Care 12, (2012).
- Morava, E. et al. Defining the Phenotype in Congenital Disorder of Glycosylation Due to ALG1 Mutations. PEDIATRICS 130, (2012).
- Dupre, T. et al. Guanosine diphosphate-mannose:GlcNAc2-PP-dolichol mannosyltransferase deficiency (congenital disorders of glycosylation type Ik): five new patients and seven novel mutations. Journal of Medical Genetics 47, (2010).
- González-Domínguez, C. A. et al. ALG1-CDG Caused by Non-functional Alternative Splicing Involving a Novel Pathogenic Complex Allele. Frontiers in Genetics 12, (2021).
- Bengtson, P. et al. Serum transferrin carrying the xeno-tetrasaccharide NeuAc-Gal-GlcNAc2 is a biomarker of ALG1-CDG. Journal of Inherited Metabolic Disease 39, (2016).
- Bieberich, E. Synthesis, Processing, and Function of N-glycans in N-glycoproteins. in (2014). doi:10.1007/978-1-4939-1154-7_3.
- Stanley, P., Taniguchi, N. & Aebi, M. N-glycans. in Essentials of Glycobiology [Internet] (eds. Varki, A. et al.) (Cold Spring Harbor Laboratory Press, 2017).
- Huffaker, T. C. & Robbins, P. W. Temperature-sensitive yeast mutants deficient in asparagine-linked glycosylation. Journal of Biological Chemistry 257, 3203–3210 (1982).
- Jaeken, J., Lefeber, D. & Matthijs, G. Clinical utility gene card for: ALG1 defective congenital disorder of glycosylation. European Journal of Human Genetics 23, (2015).
- Öncül, Ü., Kose, E. & Eminoğlu, F. T. ALG1-CDG: A Patient with a Mild Phenotype and Literature Review. Molecular Syndromology 13, (2022).
- Dhamija, R. & Chambers, C. Clinical and Molecular Characterization of ALG1-CDG. Pediatric Neurology Briefs 30, (2016).
- Nagra, S. & Dang, S. Protein Losing Enteropathy. in StatPearls [Internet] (StatPearls Publishing, 2021).
- Chang, I. J., He, M. & Lam, C. T. Congenital disorders of glycosylation. Annals of Translational Medicine 6, (2018).
- Jia, J.-X. et al. Chemo-enzymatic synthesis of the ALG1-CDG biomarker and evaluation of its immunogenicity. Bioorganic & Medicinal Chemistry Letters 30, (2020).
- Janik, A. et al. Overexpression of GDP-mannose pyrophosphorylase in Saccharomyces cerevisiae corrects defects in dolichol-linked saccharide formation and protein glycosylation. Biochimica et Biophysica Acta (BBA) - General Subjects 1621, (2003).
- Brasil, S. et al. CDG Therapies: From Bench to Bedside. International Journal of Molecular Sciences 19, (2018).