
Welcome

Finding out you or your loved one has a congenital disorder of glycosylation (CDG) can be overwhelming. These feelings are completely natural and there are many trained and specialized people available to help you or your loved one on this journey.
Learning more about CDG and the terminology can help prepare yourself and your loved ones for living with and managing CDG, as well as being able to understand and communicate needs with health care professionals.
You may have many questions around CDG and how you can help your loved one, including:
- What is CDG and how is it caused?
- How is CDG managed?
- What signs should I be aware of?
This page has been developed to help you navigate these questions, discussing the complex terms and biology surrounding CDG. You can also find additional valuable resources that have been developed by the CDG community on CDG Hub here.
What is CDG?
Congenital disorders of glycosylation (CDG) are a large group (more than 160 types) of rare inherited disorders that affect a complex process in the body called glycosylation. Glycosylation is a process where sugars are added to proteins or fats (known as lipids). Many different proteins and lipids in the body need to be attached to sugars to work properly. CDG arise from these proteins or lipids lacking the right sugars, or missing sugars altogether.
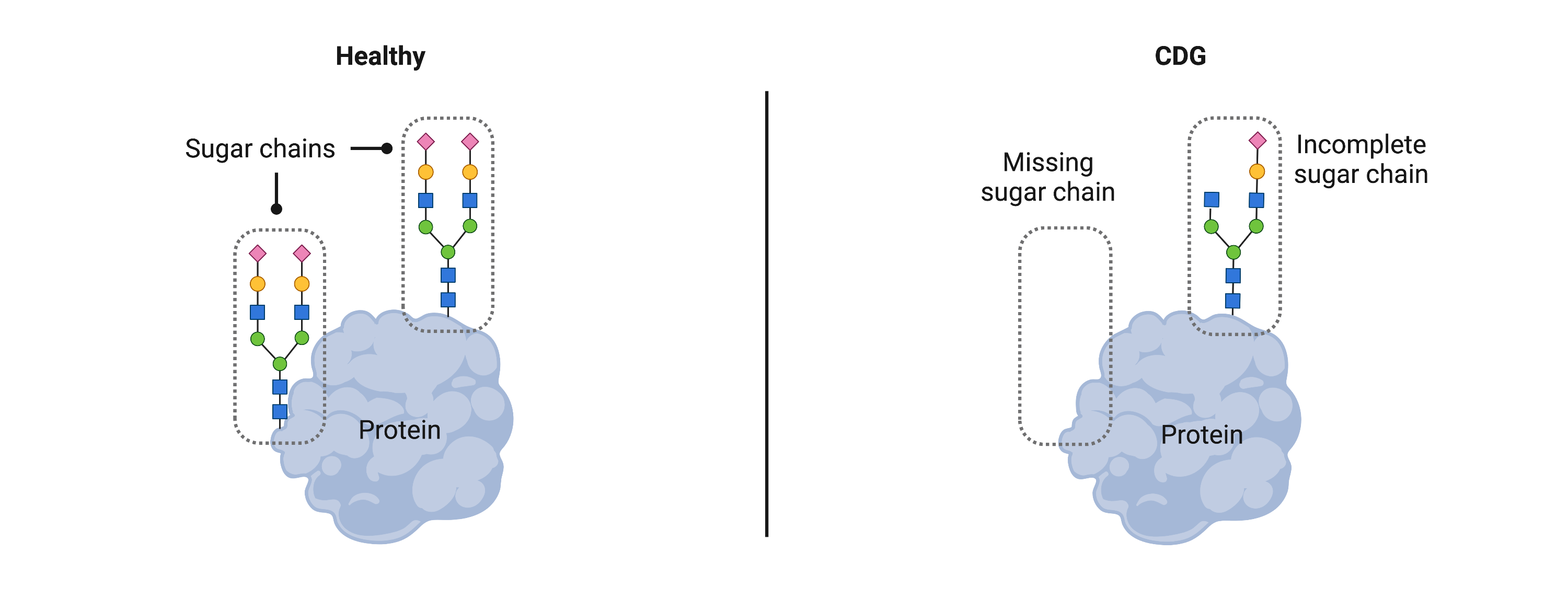
As glycosylation is needed for many proteins and lipids to work throughout the body, multiple areas of the body can be affected by CDG, causing health problems in different systems. In most cases, symptoms of CDG appear in infancy. These symptoms can range from mild to severe and can vary widely between individuals with different and even the same type of CDG. Common symptoms of CDG include intellectual disability, developmental delay, diminished muscle tone, seizures, problems with balance and coordination, growth problems, liver disease, and problems with bleeding and blood clotting. With the variability of symptoms and disease severity, CDG are typically diagnosed through a genetic test.
The following sections outline the basics of glycosylation, including the types of molecules involved and the causes of CDG.
Genes
Our cells contain structures called chromosomes that house our DNA (Figure 1). DNA is the genetic code that contains all the information cells need to generate molecules for our bodies to work. A gene is a section of DNA that instructs cells how to make a particular protein. To make a protein, DNA is converted into RNA using an enzyme (polymerase). By reading the RNA sequence, a particle (called a ribosome) can then generate a protein.
Human DNA has over 400 genes that provide instructions for making proteins that play a role in glycosylation. Changes, known as genetic mutations or pathogenic variants, in any one of these genes can cause cells to make faulty forms of proteins involved in glycosylation. As a result, sugars cannot be attached properly to proteins and lipids, resulting in CDG.
Each CDG type arises from pathogenic variants in a single gene. You may come across CDG described as being monogenetic, which means a single CDG type is caused by pathogenic variants in a single gene.
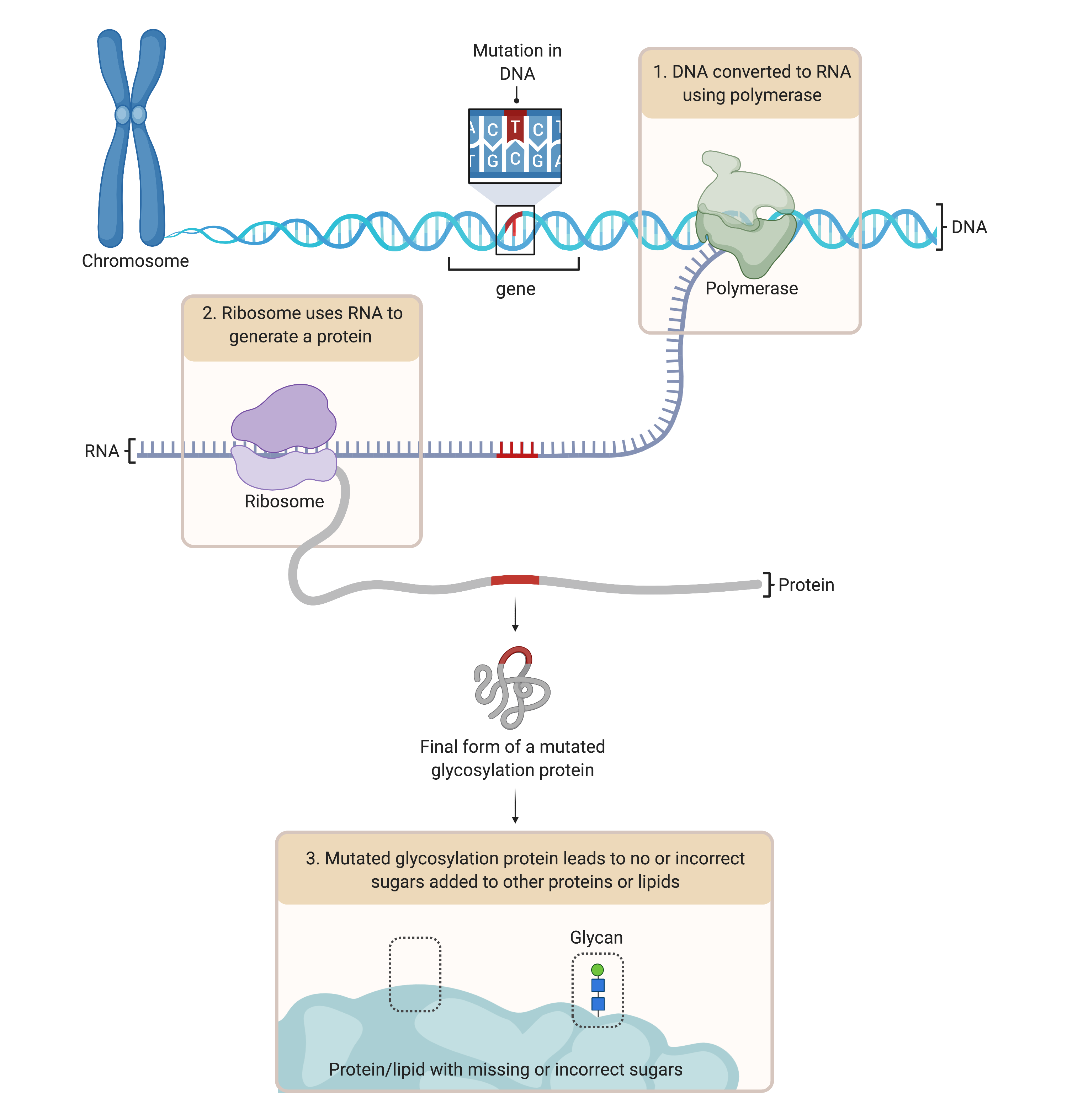
Figure 1. Overview of gene mutations leading to incorrect sugars added to proteins.
To generate proteins, DNA from a gene is converted to RNA and then the protein. CDG are caused mutations in the DNA of glycosylation genes. Here, the faulty DNA is converted to RNA, which also contains a mutation in its sequence. The faulty RNA then generates a defect in the protein, such as a glycosylation enzyme. This defective enzyme cannot work properly, leading to defects in glycosylation, such as absent sugars on a protein.
Glycosylation
Glycosylation is the process of adding sugars to proteins or lipids (Figure 2). Glycosylation can occur in different locations of the cell – depending on the type of sugar being added to the protein or lipid –and requires a variety of enzymes and other molecules.
Proteins and lipids – molecules that need glycosylation to work
Our bodies need molecules like proteins and lipids for a wide array of tasks, such as helping cells communicate with each other and helping the body fight off infections, as well as for healthy brain function and growth and development. Many proteins and lipids need sugars attached to them work properly—or, in other words, proteins and lipids need to be glycosylated.
Sugars – molecules that glycosylation proteins and lipids
The words “sugars” or “sugar building blocks” are used interchangeably and frequently in the CDG community. Sugars are not just used by the body as an energy source but are a large and diverse group of molecules.
During glycosylation, chains of sugars can be attached onto proteins or lipids, and these sugar chains are referred to as glycans. When glycans are attached to proteins, these new molecules are called glycoproteins; and when glycans are attached to lipids, these new molecules are called glycolipids. Sugars must first be ‘activated’ to be added to proteins or lipids. Simple sugars or ‘monosaccharides’ enter the cell, where they are then activated.
Endoplasmic Reticulum and Golgi –where glycosylation takes place
Glycosylation is a complex process that happens throughout the body, typically in specific compartments in cells called the endoplasmic reticulum (ER) and the Golgi Apparatus (Golgi). Activated sugars are transported from the cytoplasm the inside of these compartments.
Glycosylation often begins in the ER where sugar chains are attached to proteins or lipid-containing molecules. After glycans are attached, the glycoprotein or glycolipid will be transported to the Golgi. Here, the glycosylation process continues as glycans can be modified further, such as adding additional sugars or other types of molecules to the glycans. The glycosylated protein or lipid is then released from the Golgi and moved to a location in the body where it performs its role, which can be at the cell surface or inside or outside of the cell.
However, not all types of glycosylation (also called glycosylation pathways) follow this sequence of events. For example, many lipids and various proteins begin their glycosylation process in the Golgi.
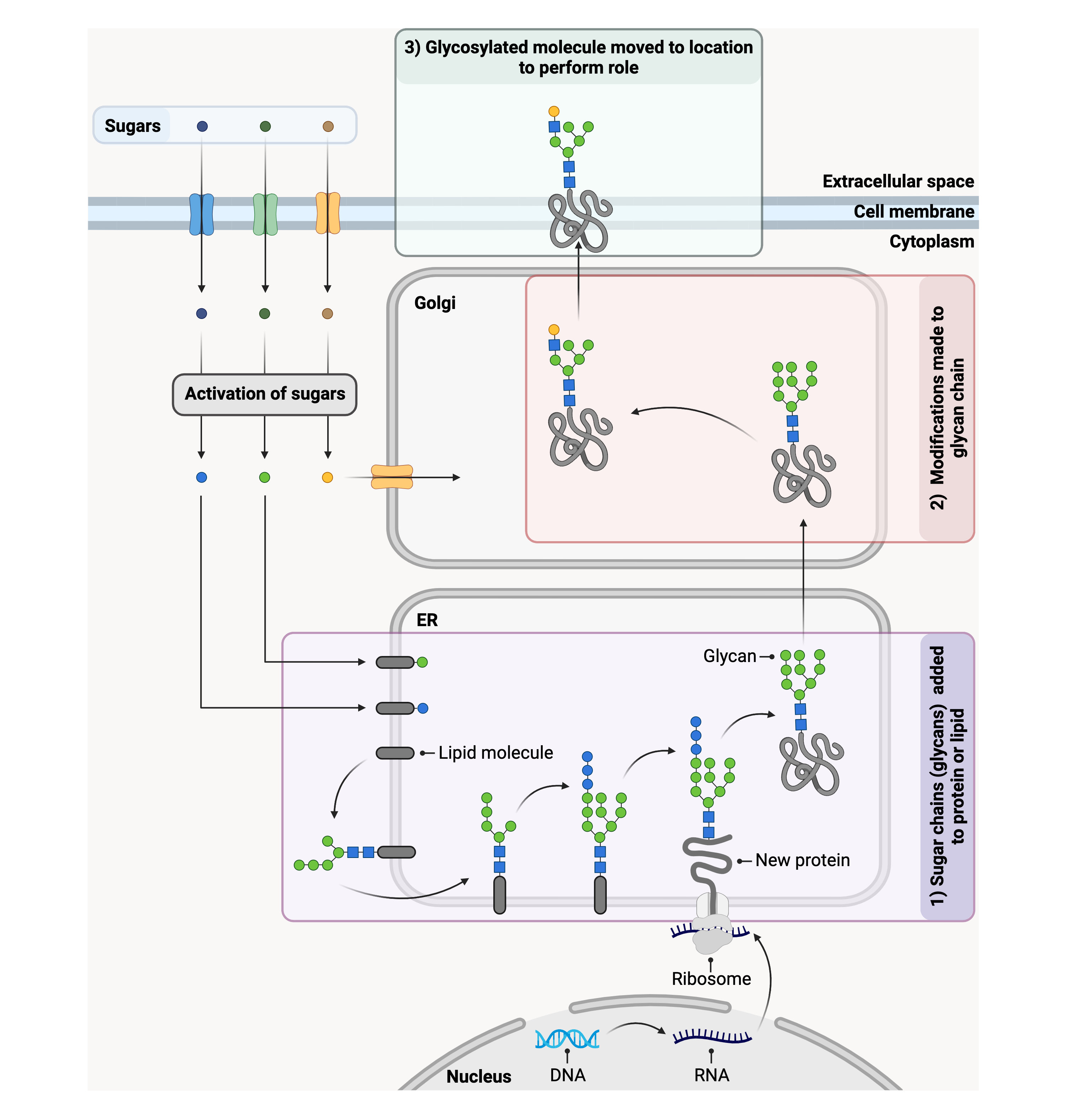
Figure 2. Overview of glycosylation of proteins in cells.
DNA is converted to RNA in the nucleus. The RNA is converted into a protein by ribosomes, which can be found on the outside of the endoplasmic reticulum (ER). Sugars chains (glycans) are attached to the protein on the inside of the ER, forming a glycosylated protein or glycoprotein. The glycoprotein is then moved to the Golgi, where the glycan can undergo further changes, such as additional sugars being added to the glycan. The glycoprotein is then moved to its final location to perform its role in the body.
Causes of CDG
CDG are caused by defects in proteins that are needed for glycosylation. Since glycosylation is a complex process relying on hundreds of proteins, especially enzymes (a type of protein), a defect in any one of these proteins could lead to CDG. These faulty proteins arise from pathogenic variants in the genes of individuals with CDG.
Inheritance
Learning how CDG arise in affected individuals can be useful for understanding the impacts and health risks for the wider family and future generations.
Inheritance refers to how genes are passed from parent to child. CDG are often inherited diseases, meaning that in most cases the pathogenic variants are inherited from a child’s parents. Cells typically contain two copies of each gene: one copy that is inherited from the mother and one copy that is inherited from the father. This is called autosomal inheritance.
Some genes are located on the sex chromosomes, X and Y. Females have two X chromosomes (XX) and males have one X and one Y chromosome (XY). Pathogenic variants in genes found on the X chromosome can cause CDG through X-linked inheritance.
Depending on the CDG type, one or both copies of a gene must be faulty to cause disease. In most types of CDG, a child’s parents carry the pathogenic variant but do not have symptoms of CDG.
There are four main types of inheritance that are known to cause CDG (Figure 3):
- Autosomal Recessive – where both copies of a gene need to contain pathogenic variants to cause disease, meaning an individual must have two faulty copies of the gene in their cells. Most types of CDG are inherited in an autosomal recessive pattern.
- Autosomal Dominant – where only one faulty copy of a gene is needed to cause disease. The faulty gene can be inherited from either parent.
- X-linked Dominant – for X-linked dominant inheritance, females require both gene copies in their pair of X chromosomes to carry a pathogenic variant. As males only have one X chromosome, disease occurs when the single X chromosome contains a pathogenic variant of the gene.
- X-linked Recessive– this form of inheritance typically occurs only in males. One copy of a faulty gene found on the X chromosome is inherited from the mother.
Some pathogenic variants that cause CDG can develop in genes spontaneously, meaning these pathogenic variants are only found in the genes of an affected individual. These variants are described as de novo variants8.
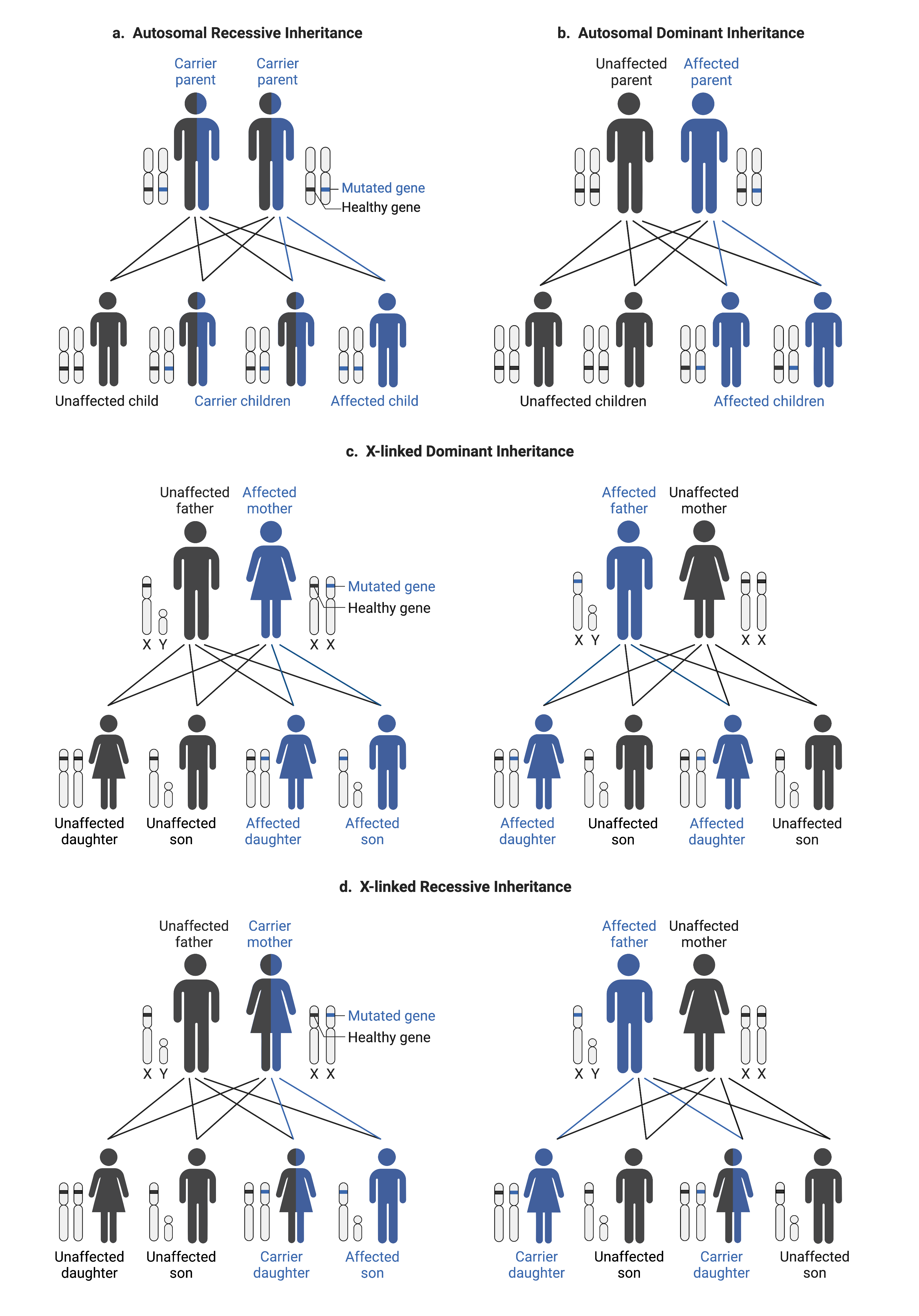
Figure 3. Potential ways CDG can be inherited, including autosomal recessive (a) or dominant (b) and x-linked dominant (c) or recessive (d).
Classification of CDG Types
There are currently over 170 known types of CDG, with new types discovered regularly. As more CDG have been identified, new ways of grouping and naming these disorders have emerged.
CDG Naming
Traditionally, CDG were classified into two groups: type I or type II, depending on results from a blood test. The CDG type was then named CDG-I or CDG-II, depending on the group it belonged to. Each new disorder discovered in a group was then given a letter of the alphabet (in order of discovery), such as CDG-Ia and CDG-Ib.
However, this classification and naming system became outdated as more CDG types were discovered because many did not fit into either of these groups. Consequently, a new naming system was developed, where each CDG type is named after the mutated gene that is responsible for causing the disorder (i.e. GENE-CDG). For example, PMM2-CDG, the most common type of CDG, is caused by mutations in the PMM2 gene.
CDG Classification
CDG can be broadly classified into five groups, depending on which glycosylation pathways are affected.
Disorders of N-linked Protein Glycosylation
These CDG are caused be defects in the N-glycosylation pathway, one of the main types of glycosylation of proteins. Disorders of N-linked protein glycosylation are the most common form of CDG.
Disorders of O-linked Protein Glycosylation
CDG belonging to this group are caused by defects in the O-glycosylation pathway which is another main type of glycosylation of proteins. Some CDG in this group can also be classified as forms of muscular dystrophy, which are called dystroglycanopathies as they have abnormal glycans attached to a protein called alpha-dystroglycan.
Disorders of GPI Anchor Biosynthesis and Lipid Glycosylation
These disorders affect the attachment of sugars to lipids or molecules containing lipids, such as GPI-anchors which help attach proteins to the cell surface.
Disorders of Multiple Glycosylation Pathways
These CDG are caused by defects in genes that play a role in multiple glycosylation pathways, such as different types of protein and lipid glycosylation.
Disorders of Deglycosylation
Disorders of degylcosylation are caused by defects in the removal of sugar chains from proteins (deglycosylation) and their subsequent breakdown. CDG belonging to this group are called congenital disorders of deglycosylation (CDDG) and are named according to the affect gene, followed by -CDDG. Currently, only NGLY1-CDDG and MAN2C1-CDDG belong to this category.
Signs and Symptoms
Symptoms of CDG often appear in infancy although for some CDG types, symptoms may not appear until adulthood. CDG can affect multiple body systems, where symptoms vary between different types of CDG and within the same type of CDG. Consequently, someone with CDG may not have all the symptoms that are associated with their CDG type, as specific symptoms and potential complications depend on which systems are affected. These symptoms can also vary in severity and can appear from infancy.
Symptoms in infants and children may include:
- Growth problems – feeding difficulties, slow weight gain (failure to thrive), delays in getting taller
- Development delays – delays in reaching developmental milestones, such as talking or walking later than others
- Neurological problems – decreased muscle tone (hypotonia), seizures, poor balance and coordination (ataxia), stroke-like episodes
- Eye problems – misaligned or crossed eyes (strabismus), vision problems
- Liver problems – liver disease resulting in various issues like weight loss, inability to think clearly, yellowing of skin
- Heart problems – fluid accumulation around the heart, thickening and stiffening of the heart muscle (cardiomyopathy)
- Bleeding problems – increased bleeding because of blood clotting problems, increased risk of blood clot formation (thrombosis)
- Hormonal problems – low blood sugar due to high levels of a hormone called insulin (the condition is also called hypoglycemia), tiredness and other related symptoms due to decreased levels of a thyroid hormone (called hypothyroidism), reduced growth due to a lack of growth hormone production
- Bone problems – curving of the spine, and other joint problems
- Immunologic problems – severe or long-lasting infections
- Gut problems – chronic diarrhea, throwing up
- Skin problems – loose skin (cutis laxa), rash, scaly skin, peeling of skin, fat pads
Symptoms in adulthood may include:
- Intellectual disability
- Problems with balance, coordination, and speech
- Slurred speech (dysarthria)
- No puberty in girls
- Joint problems
- Liver disease
- Vision problems (retinitis pigmentosa)
Diagnosis
Since many symptoms of CDG can appear in the first few months of life, CDG is often diagnosed in infancy. However, some types of CDG, such as MPI-CDG, may be diagnosed in adulthood. Diagnosis usually begins with your health care provider performing a thorough physical examination of someone suspected of having CDG, taking family medical history into consideration. Because symptoms of CDG are similar to other conditions, individuals with CDG are often mistakenly diagnosed. Some may be misdiagnosed with cerebral palsy or other neurological or genetic conditions. It is important that these conditions are ruled out during diagnosis.
Your health care provider may perform a series of tests during CDG diagnosis, such as:
- Transferrin test
- Blood tests
- Genetic tests
Transferrin Test
A transferrin test may also be called transferrin isoform analysis, transferrin analysis, or carbohydrate-deficient transferrin (CDT) analysis. This test measures missing or incorrect glycans on a blood protein called transferrin, which helps move iron around the body (Figure 4). This test is commonly performed during CDG diagnosis, but it can only detect certain groups of CDG and cannot determine the type of CDG.
Figure 4. Transferrin with healthy glycans and abnormal glycans. Analysis of transferrin in the blood is a screening test commonly used to diagnose CDG.
Blood Tests
CDG can cause many problems in other proteins in the blood stream, such as elevated liver enzymes (AST and ALT), defects in blood clotting factors (coagulation) and abnormal hormone levels. Consequently, blood tests are often performed to identify whether or not someone with CDG has abnormal levels of various proteins in their blood.
Genetic Testing
Genetic testing is the most reliable way to diagnose CDG. It can determine the specific CDG type by identifying the affected gene.
Management
With no cure for CDG, treatment for most CDG types is primarily supportive and is directed towards managing the specific symptoms that are present in each individual, preventing complications and enhancing their quality of life.
CDG management depends on the CDG type, which body systems are affected, as well as your loved one’s symptoms, age, and other health conditions.

Find a CDG Expert
Finding medical specialists familiar with CDG is important for developing an effective management strategy. Meeting with an expert can also offer guidance and help you find the right support for your journey. Visit the Clinicians and Centres page to find CDG medical professionals and clinics near you.
Team of Healthcare Professionals
Symptoms may vary depending on the type of CDG. As CDG tends to affect many body systems, it is important to have a healthcare team with multiple specialists. They can not only help treat symptoms of CDG but also work with you to identify ways to improve the quality of life for you or your loved one.
- Primary care pediatricians specialize in providing medical care to children, including routine check-ups.
- Nurse practitioners and special nurses are nurses that have undergone specialized training and provide support when your loved one must stay in hospital.
- Occupational therapists (OTs) specialize in identifying and providing treatment plans for problems surrounding everyday activities, such as eating, getting dressed, going to school, and washing, helping your loved one safely develop and refine these skills.
- Physical therapists (PTs) focus specifically on physical disabilities, and provide treatment plans, activities, and goals to help develop physical skills to enhance your loved one’s quality of life.
- Speech language pathologists (SLPs) assess speech or feeding problems and provide speech therapy, where they will work through exercises tailored to the needs of your loved one to help them improve their speech, chewing, swallowing, and drinking.
- Gastroenterologists specialize in digestive health problems and identify gut defects, such as reduced nutrient absorption and diarrhea, and provide treatment options.
- Registered dietitians work with gastroenterologists to further develop digestive treatment options and treatments for nutritional deficiencies.
- Ophthalmologists will help diagnose and treat vision problems.
- Audiologists help diagnose specific problems associated with hearing and/or balance and provide management strategies.
- Ear, nose & throat (ENT) specialists perform surgeries that may be required to improve hearing or balance, in addition to helping provide diagnoses.
- Cardiologists specialize in identifying heart defects that can occur in some individuals with CDG and can also provide treatment options.
- Endocrinologists identify abnormal hormone, growth, and blood glucose levels and will provide treatment options to restore hormone levels and treat symptoms associated with hormone imbalances.
- Genetic counsellors are involved with genetic testing and diagnosis and provide a safe, confidential, and accepting environment for you and/or your family to share personal information, while helping making treatment decisions.
- Social workers support you and your loved ones, as well as identifying resources to help with financial and care needs.
Potential Complications Associated with CDG
Individuals with CDG may be at risk of complications, depending on their CDG type and affected systems. Complications can have unique signs, which you can identify and seek emergency medical care—for more information, speak to your health provider or see resources such as the Mayo Clinic Patient Education booklets. Details of some of these complications, and what they are, are listed below (obtained from Mayo Clinic Booklet).
Blood Clot Blocking a Blood Vessel (Thrombosis)
A blood clot is a gel-like mass (made from blood cells and proteins) that the body produces to stop bleeding after injury. Blood clots are usually broken down by the body but can be dangerous if the clot travels to the brain or heart as it can block blood flow around these organs.
Seizures
A seizure is an uncontrolled surge of electrical activity in the brain, which can alter how people behave, move, and feel, as well as a person’s consciousness.
Infections
Infections are caused by disease-causing microorganisms invading and growing in the body.
Stroke-Like Episodes
Stroke-like episodes occur when there is reduced blood flow to the brain and individuals can display symptoms that resemble a stroke.
Uncontrolled Bleeding
Uncontrolled bleeding occurs when the blood does not clot during bleeding.
Considerations for Families and Caregivers
If you are a parent, family member or caregiver of someone with CDG, knowing their medical history will put you in a good position to care and advocate for them. If their healthcare provider has told you that your loved one is prone to certain problems, keeping an eye out for related symptoms and ensuring they have regular check-ups can help manage potential complications.
As a caregiver, balancing your time between caring for your loved one, yourself, and others in your family may feel challenging. You may have feelings of being physically and emotionally overwhelmed while caring for your loved one with CDG. These feelings are completely natural, and you can seek support from your healthcare provider, by joining support groups, and asking or accepting help from friends and family.
The CDG CARE Family Support Network connects patients, families and caregivers around the world to provide support, share resources and improve the lives of families living with CDG.
Treatments
CDG treatments largely focus on dietary supplements (such as different types of sugars) and, in some cases, organ transplants. Additional types of treatments are also being developed in clinical trials—which are studies that are used to generate medical information on humans, such as the effects of new treatments.
Non-dietary treatments in pre-clinical studies or clinical trials include repurposed drugs, pharmacological chaperones and gene therapy.
You can learn more about therapies currently in development for CDG on the World CDG Organization website here.
Dietary Supplementation
Dietary supplementation is one of the most widely applied treatment strategies for CDG. The goal of dietary supplementation is to provide molecules that are needed to restore the normal glycosylation process in affected individuals. For example, dietary supplements can include the simple sugars or sugar building blocks (called monosaccharides) needed for cells to build glycans during glycosylation.
Organ Transplantation
Organ transplantation, where an individual receives an organ from another body, has been a successful treatment option in cases with severe and progressive liver disease.
Drug Repurposing
Drug repurposing (repurposed drugs or drug repositioning) is where an existing drug is used to treat different diseases to those that it was originally intended for. Several repurposed drugs are currently under investigation in clinical trials for the treatment of PMM2-CDG (epalrestat and acetazolamide) and in early stages of study for SRD5A3-CDG, PIGA-CDG and MAN1B1-CDG.
Pharmacological Chaperones
Pharmacological chaperones are small molecules that stick to faulty proteins, restoring their ability to work properly. This therapeutic approach is currently under investigation to treat PMM2-CDG.
Gene Therapy
Gene therapy can involve replacing a faulty form of a gene with a healthy form of the gene in affected individuals, allowing these individuals to generate healthy proteins. Gene therapy is in early stages of study for PIGA-CDG and SRD5A3-CDG.
Prognosis
As CDG encompasses a wide variety of symptoms which range in their severity, prognosis varies greatly between different types of CDG and even between individuals with the same CDG type. It is important to talk to your physician and medical team to better understand your loved one’s specific symptoms, unique needs and prognosis.

Many individuals with CDG have disabilities that begin at a young age. Children with CDG often experience delays in acquiring new skills as they grow and develop, such as gross and fine motor skills, speech, language as well as social and cognitive skills. Some children may have cognitive impairments or learning disorders. Regular visits with physical, occupational and speech therapists on your healthcare team can help in improving your loved one’s skills and mobility.
Some children with CDG have serious medical problems which can be life-threatening and may require frequent or lengthy hospital stays. However, the frequency of critical episodes typically decreases as children grow up. Although most CDG types do not have specific treatments available, many health concerns and symptoms in individuals with CDG are treatable. Regular check-ups by your medical team, preventative medicines and awareness of potential complications can reduce the risk of serious complications in CDG.
In adulthood, symptoms of CDG can be non-progressive. However, most adults with CDG do not reach independence and require a supportive environment. Ongoing research is focused on better understanding how CDG progresses in order to identify ways to improve the quality of life for people living with CDG.
Clinical Studies
Finding effective therapies for the many types of CDG will require research involving various types of clinical studies. Clinical studies are studies that generate medical information on humans, such as how well a drug works at treating a disease or the side effects of a drug or how a disease progresses. Clinical studies can be divided into clinical trials (or interventional studies) and observational studies.
Learn more about ongoing clinical studies on CDG on the World CDG website here.
Clinical Trials
Clinical trials (or interventional studies) assess clinical ‘interventions’, such as a drug, procedure or behavioural change, in participants, which are assigned by the study researcher. Clinical trials can provide information on the safety, effects, and effectiveness of an intervention. These studies can be split into different phases (1–4), outlined by the FDA, which are determined based on factors such as the study aim and participants.
To learn about active CDG clinical trials visit the Clinical Trials page.
Access educational materials for CDG families on clinical trials that have been developed by World CDG Organization here.
Observational Studies
Observational studies monitor how a disease progresses and the health outcomes. Participants can receive clinical interventions in the study, but these are not assigned by the researcher.
A natural history study is a type of observational study that gathers information on patients to understand how a disease develops and progresses, as well as treatment options. This allows for a better understanding of the disease, which is especially beneficial in the case of rare inherited disorders like CDG as so little is known about these diseases. Currently, a large 5-year natural history study on all types of CDG is being conducted by the Frontiers in CDG Consortium. Learn more about this study here.
Research
Ongoing work from researchers around the globe is helping us better understand CDG and is critical to improving care, identifying better diagnostic techniques and developing effective treatment options. You can learn about CDG researchers from around the world on the Researchers page and find out about research opportunities to participate in here.