
Lay Summary
PMM2-CDG, formerly known as CDG-Ia, is a rare inherited condition that affects many systems in the body. It is the most frequent congenital disorder of glycosylation (CDG) with more than 1000 patients reported in the medical literature to date. PMM2-CDG is typically classified as a disorder of N-linked protein glycosylation. PMM2-CDG is caused when an individual has mutations in both copies of the PMM2 gene, which provides instructions for making an enzyme that is required for generating molecules that donate mannose sugars onto growing sugar chains during N-glycosylation. Mutations in the PMM2 gene cause some proteins to have incomplete or absent sugar chains. Symptoms of PMM2-CDG usually begin at infancy, affect multiple systems, and can change over the lifespan of the affected individual. Neurological abnormalities, including developmental delay, low muscle tone, epilepsy, and problems with coordination and balance are common. Inverted nipples and abnormal fat pads are common in children but usually resolve over time. Problems with heart and spine formation may require surgery to address. Several screening tests are available for PMM2-CDG, but a definitive diagnosis is achieved through genetic testing. There are currently no approved treatments for PMM2-CDG, but two drugs are being explored in clinical trials. Treatment is focused on the management of specific symptoms and preventing disease complications.
Overview
Phosphomannomutase 2 congenital disorder of glycosylation (PMM2-CDG) is a rare autosomal recessive genetic disorder that arises from defects in the PMM2 (phosphomannomutase 2) gene 1,2. The PMM2 gene encodes an enzyme (PMM2) responsible for converting mannose-6-phosphate to mannose-1-phosphate. Mannose-1-phosphate is a precursor for GDP-mannose, which is the mannose donor used in the construction of mannose-containing sugar chains (glycans) in the cytoplasm. GDP-mannose is also used in the biosynthesis of dolichol-phosphate-mannose (Dol-P-mannose), which is the donor of mannose sugars inside the endoplasmic reticulum (ER). These mannose donors are crucial for the early steps of protein N-glycosylation, and deficiency in PMM2 results in insufficient N-glycosylation of glycoproteins.
The first two patients with a PMM2-CDG phenotype were reported in 19803,4. Later studies in the 1990s linked this phenotype to mutations in PMM2-CDG1,5. There are over 1000 confirmed cases to date, making it the most common congenital disorder of glycosylation1,6,7. Due to underdiagnosis, it is estimated that the prevalence of PMM2-CDG could be as high as 1:20,000 (based on European data)8,9. However, it is clear that there are regional differences in PMM2-CDG frequency, and carrier frequency for different pathogenic PMM2 gene variants also varies by region8,10,11. Symptoms are highly variable but typically begin at infancy. The clinical characteristics of PMM2-CDG include a variety of neurological symptoms, a small cerebellum, intellectual disability, gastrointestinal and feeding problems, cardiac issues, clotting disorders, low bone density and scoliosis, abnormal fat pad distribution in childhood, and progressive peripheral neuropathy and visual decline in adulthood2,12. A diagnosis can be determined through transferrin analysis and direct assessment of PMM2 enzymatic activity, but definitive diagnosis is often achieved through genetic testing. Dietary mannose therapy has been explored as an option for treating PMM2-CDG, but its efficacy is controversial13–15. Acetazolamide is in a phase 3 clinical trial for the treatment of neurological symptoms and Epalrestat is in a phase 2 clinical trial to examine its ability to indirectly increase PMM2 activity, but no cure is currently available16,17.
Synonyms
- CDG-Ia, CDG-1A
- CDG IA, CDG1A
- Carbohydrate-deficient glycoprotein syndrome 1a (CDGS 1a)
- Congenital disorder of glycosylation type 1a, Congenital disorder of glycosylation type Ia
- Phosphomannomutase 2 deficiency
- PMM2 Deficiency
- Carbohydrate-deficient glycoprotein syndrome type Ia, Carbohydrate-deficient glycoprotein syndrome type 1a
- CDG syndrome type Ia, CDG syndrome type 1a
- Jaeken Syndrome (aka Carbohydrate deficient glycoprotein syndrome type 1): an antiquated name for a syndrome that encompasses several CDG, including PMM2-CDG
Inheritance
PMM2-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent1,2. In several cases, PMM2-CDG has been caused when two copies of a defective allele are inherited from the same parent (typically the mother), a rare process known as uniparental disomy18,19.
Gene Function
The PMM2 gene encodes a mutase enzyme, phosphomannomutase 2 (PMM2). Mutases are enzymes that facilitate the movement of part of a molecule from one place to another on the same molecule. PMM2 is located in the cytoplasm where it has a role in the generation of mannose-1-phosphate from mannose-6-phosphate (Figure 1).
The PMM2 enzyme catalyzes the conversion of mannose-6-phosphate to mannose-1-phosphate. Mannose-1-phosphate is the precursor of the activated sugar GDP-mannose. Activated sugars are also called glycosyl donors or sugar donors because they enable sugars to be added onto other molecules. This is particularly important for N-glycosylation of proteins, which involves the attachment of a mannose-rich glycan to the amino acid asparagine on a protein. Overall, PMM2 activity ensures that there is enough mannose-1-phosphate to support the generation of the mannose glycosyl donors used in N-glycosylation.
Monosaccharide Inteconversion
The monosaccharide mannose is an important component of glycans that are attached to various proteins and lipids. Before mannose can be incorporated into glycans, it must be converted to GDP-mannose in the cytoplasm. Several steps are required to generate GDP-mannose from mannose, including the conversion of mannose-6-phosphate (M6P) to mannose-1-phosphate (M1P). GDP-mannose is directly synthesized from mannose-1-phosphate, and the PMM2 gene encodes the isomerase enzyme (PMM2) responsible for converting mannose-6-phosphate to mannose-1-phosphate (Figure 1). GDP-mannose is also a precursor for Dol-P-mannose (Dol-P-Man), the mannose donor used inside the ER.
The first step of N-glycosylation is the building of a mannose-rich lipid-linked oligosaccharide (LLO) comprised of a 14-sugar oligosaccharide and dolichol pyrophosphate (Dol-PP). The oligosaccharide component, also referred to as the oligosaccharide precursor, is subsequently transferred en bloc to an asparagine (Asn; N) residue of a protein inside the ER. The synthesis of the LLO occurs through a stepwise process that occurs both in the cytoplasm and inside the ER, and thus requires both GDP-mannose and Dol-P-mannose as glycosyl donors.
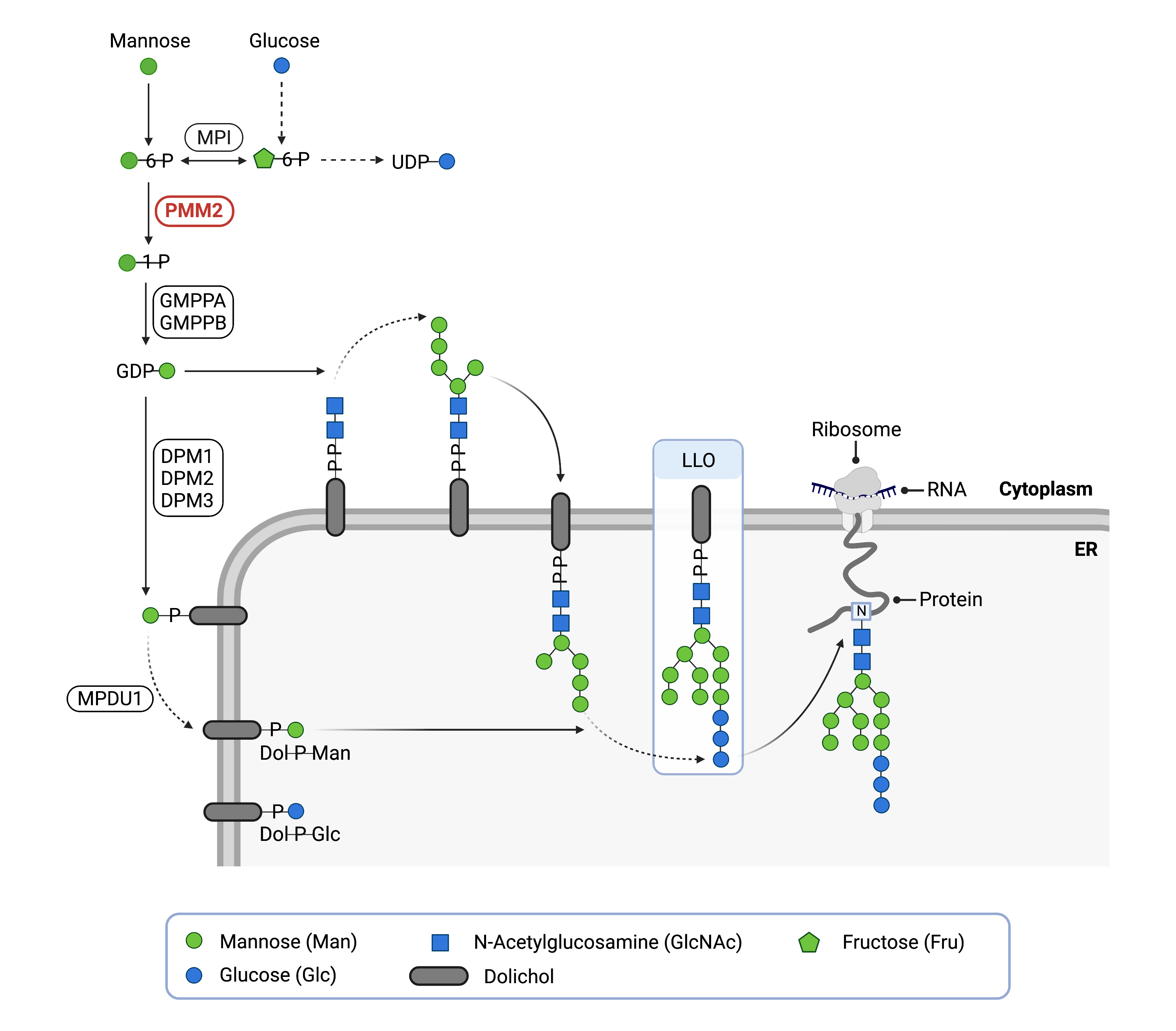
Figure 1. Role of PMM2 in N-glycosylation.
PMM2 converts mannose-6-phosphate into mannose-1-phosphate, which is needed to generate activated forms of mannose (GDP-mannose and Dol-P-mannose) for N-glycosylation.
Disease Mechanism
Mutations in the PMM2 gene lead to the production of an abnormal enzyme with reduced activity, leading to lower levels of mannose-1-phosphate. In turn, this decreases the amount of mannose-based glycosyl donors available for use in the synthesis of glycans. Insufficient levels of GDP-mannose and Dol-P-mannose resulting from reduced PMM2 activity is particularly problematic for the N-glycosylation of proteins. Synthesis of the mannose-rich LLO is impaired in PMM2 deficiency, causing N-glycosylated proteins to be insufficiently or incorrectly glycosylated (hypoglycosylation). Incorrect glycosylation of N-glycoproteins can cause a cascade of errors that contribute to the PMM2 CDG pathology. For example, misglycosylated matrix metalloprotein 2 and proconvertase result in altered processing of N-cadherin, a protein important for cell adhesion and normal tissue development20.
Reduced PMM2 activity also results in an accumulation of the enzyme’s substrate, mannose-6-phosphate, which has an additional role as a regulator of LLO destruction21,22. Excess mannose-6-phosphate causes decomposition of the LLO, further compounding the low levels of LLO synthesis that result from the decreased concentration of mannose-based glycosyl donors in PMM2 CDG.
Mutations
At least 118 mutations have been reported in the PMM2 gene, including 93 missense mutations, 5 nonsense mutations, 7 frameshift mutations, and 10 splicing defects. Variants resulting in the loss of exon 3 and exon 8 have also been reported23,24. The p.R141H mutation is most common and is present in 40% of patients (75% of patients of Caucasian origin)9,25,26. In Europe, other common mutations are p.F119L, p.V231M and p.P113L; the most commonly identified genotype is p.R141H/p.F119L12.
Due to the number of identified mutations and the variability of phenotype observed between patients with the same genotype, it is difficult to correlate disease severity with a particular genotype. However, the p.R141H allele, present in the most common genotype (p.R141H/p.F119L), is typically associated with more severe disease and all patients with this mutation are heterozygous, as the homozygous genotype is incompatible with life8,9,25,26. In some cases, the p.R141H mutation may display a milder phenotype. Particularly, the p.R141H/p.T226S, p.R141H/p.I132T, and p.R141H/p.Q139K genotypes appear to be associated with milder presentations in PMM2-CDG27. Some less common genotypes are enriched in certain populations and are associated with less severe disease; p.L32R is more common in Italy and is associated with a mild neurological phenotype28.
It has been suggested that disease severity is correlated with the amount of remaining PMM2 activity; mutations that lead to greater loss of PMM2 function result in more severe disease. Complete loss of PMM2 function (less than 3-7% of normal baseline activity) is fatal17. Mutations in the C-terminus of the protein, including p.H218L, p.T237M, and p.C241S may be associated with a milder phenotype29,30. As well, p.V231M/p.R239W and p.I120T/p.G228C genotypes have caused mild PMM2-CDG with normal cognitive development31. Conversely, mutations in the dimerization domain, including p.P113L and p.F119L, result in a worse clinical course32.
A mutation in the PMM2 promoter region has also been reported to cause a syndrome characterized by polycystic kidneys and hyperinsulemic hypoglycemia, but affected individuals have an otherwise normal phenotype33.
Signs & Symptoms
Clinical Presentation
Patients with PMM2-CDG may display a wide range of symptoms with varying severity. In the most extreme cases, PMM2-CDG can cause accumulation of fluid in the fetus and prenatal mortality. Conversely, asymptomatic PMM2-CDG has also been reported34. Despite a wide range of possible symptoms, most PMM2-CDG patients have a clinical presentation that can be divided into three stages: infancy, childhood, and adulthood. Some signs and symptoms resolve over time, while others may worsen or remain unchanged.
Infancy
The infantile presentation is typically early onset and associated with feeding problems and failure to thrive, neurological symptoms including seizures and low muscle tone, abnormal fat pad distribution and inverted nipples, issues with multiple organ systems, ophthalmologic manifestations, and abnormal facial features.
- Neurologic – low muscle tone (hypotonia), poor reflexes, developmental delay, and cerebellar atrophy. Seizures may start between 5-53 months (average of 17 months)
- Ophthalmologic – crossed or misaligned eyes (strabismus), roving and/or rapid and uncontrollable eye movements (nystagmus), nearsightedness, decreased peripheral and night vision (retinitis pigmentosa)
- Cardiac – fluid buildup in the space around the heart (pericardial effusion), and potentially fatal cardiomyopathy and structural problems
- Feeding/Growth – failure to thrive often due to feeding problems and vomiting
- Abnormal Features – short nose, large/protruding ears, prominent forehead, and a long philtrum. Inverted nipples and subcutaneous fat pads above the buttocks and in the genital region that spontaneously resolve
Childhood
This stage occurs between the ages of 3-10 and is characterized by many of the same symptoms of the early childhood stage. IQ is usually between 40-70 with delayed attainment of developmental skills and language. Unassisted walking is rare and musculoskeletal problems may become more noticeable. Patients typically have a cheerful demeanor.
- Neurologic – low muscle tone, poor reflexes, developmental delay, delayed speech, slow attainment of fine motor skills, cerebellar atrophy, seizures, and stroke-like episodes. Progressively reduced sensation and weakness in arms and legs (peripheral neuropathy) may begin during this stage
- Ophthalmologic – crossed or misaligned eyes, abnormal eye movements, nearsightedness, and potentially worsening peripheral and night vision (retinitis pigmentosa)
- Musculoskeletal – low bone density, scoliosis, and abnormal development of bones and muscles in the torso
- Feeding/Growth – failure to thrive
- Abnormal Features – short nose, large/protruding ears, prominent forehead, and a long philtrum
Adulthood
Intellectual disability in adult patients is usually stable, with average IQs between 40-70. Adults are usually unable to walk and issues with the structure of their bones and muscles may become severe.
- Neurologic – low muscle tone, poor reflexes, difficulty speaking, poor coordination (ataxia), cerebellar atrophy, seizures, and stroke-like episodes. Progressive peripheral neuropathy is common but variable in severity
- Ophthalmologic – misaligned eyes, abnormal eye movements, progressive nearsightedness, and worsening peripheral and night vision (retinitis pigmentosa). Cataracts may also appear
- Musculoskeletal – low bone density leading to osteoporosis, worsening scoliosis and problems with the structure of the torso
- Abnormal Features – coarse facial features
Biochemical Abnormalties
Biochemical abnormalities frequently observed in individuals with PMM2-CDG include elevated liver enzymes (transaminases) and blood clotting (coagulation) abnormalities.12 Low blood sugar (hypoglycemia), hypothyroidism as evidenced by elevated thyroid stimulation hormone (TSH), low serum albumin, low cholesterol (hypocholesterolemia), and/or excess proteins and amino acids in the urine (proteinuria and aminoaciduria) is also seen in some individuals. In adolescence and adulthood, most females with PMM2-CDG do not produce the hormones involved in sexual development and therefore do not go through puberty. Males may have low testosterone and sex-hormone binding globulin but otherwise go through normal changes during puberty.
Diagnosis
PMM2-CDG should be suspected in any multisystem disorder, especially when developmental delay, hypotonia, and neurological symptoms are present. Screening in suspected patients typically begins with a blood test to analyze serum transferrin isoforms12. Direct assessment of PMM2 enzymatic activity may also be used and is often performed after a transferrin analysis displays a type I pattern35–37. However, analysis of transferrin isoforms is unreliable in the first three weeks of life, can appear normal or near-normal and can spontaneously normalize later in life12,13,38. Thus, genetic testing is the only way to definitively diagnose PMM2-CDG.
Transferrin Analysis
Individuals with PMM2-CDG show a characteristic type I pattern by transferrin isoelectric focusing (TIEF) or mass spectrometry analysis of transferrin39–41. Type I patterns are observed in CDG that arise due to defects in LLO assembly and are characterized by a decrease in tetrasialo-transferrin and an increase in di-sialo and a-sialo transferrin isoforms.
PMM2 Enzyme Activity
Direct analysis of PMM2 activity can be measured in patient fibroblasts or leukocytes (white blood cells). Measuring activity in leukocytes is preferred as fibroblasts may demonstrate higher residual PMM2 activity, potentially masking PMM2-CDG diagnosis36. In PMM2-CDG, PMM2 activity is less than 50% of normal baseline17.
Biomarkers
Sorbitol
Increased sorbitol levels have been linked to peripheral neuropathy in diabetes, and more recently it has been identified as a clinical biomarker for PMM2-CDG, with increased urinary sorbitol levels correlating to more severe disease42.
Classification
PMM2-CDG is classified as a disorder of N-linked protein glycosylation. More specifically, it is a disorder of monosaccharide interconversion.
Under the former CDG classification system, PMM2-CDG is classified as a Type I CDG, which arise due to defects in the synthesis of N-glycoproteins that occur before the glycan is transferred from the LLO onto the protein.
Prognosis
Disease outcomes vary depending on the severity of the illness. Milder forms of PMM2-CDG can involve neurological abnormalities in infancy with slow weight gain (faltering growth), crossed eyes (strabismus), developmental delay, abnormal cerebellar, and hepatopathy, along with neuropathy and retinitis pigmentosa developing in the first 20 years of life. Severe forms of PMM2-CDG involve severe neurological abnormalities along with additional symptoms, such as faltering growth, liver disease, organ failure, vomiting and renal cysts20. Mortality is approximately 20% in the first years of life due to infections, liver insufficiency or cardiomyopathy. Individuals with mild illness have been reported in the medical literature in their sixties and seventies, with instances of spontaneous improvement13,28.
Management
Management of PMM2 requires a multidisciplinary team and may include combinations of physical therapy, occupational therapy, oral motor and speech therapy, ophthalmological intervention, cardiological intervention, psychiatry, and palliative measures.
Seizure and Stroke-like Episodes
Seizures (tonic-clonic and partial) are usually managed by single antiepileptic drugs7. Full recovery from stroke-like episodes (SLE) may take between 1 hour and 6 months and are typically treated using antiepileptic drugs, in particular the benzodiazepines midazolam and lorazepam. It has been recommended that anticoagulants be considered in the acute phase of every case of SLE7.
Cardiac Issues
Treatment of cardiac issues should be guided by cardiologists based on the individual needs of the patient. Surgery may be required to address structural problems with the heart. Nutritional and hydration support is often used to treat cardiomyopathy resulting from dehydration. Pericardial effusion may be improved using steroids and salicylic acid43.
Coagulopathies
Coagulation studies are typically conducted on a yearly basis and before any surgery or invasive procedure7. The guidelines of the American College of Chest Physicians have been recommended to manage the formation of blood clots7,44,45.
Feeding Difficulties
Feeding difficulties, especially common in early life, may require the use of feeding supports, e.g. feeding through a tube in the nose (nasogastric feeding) or through a tube directly into the stomach (gastrostomy feeding)7. Regurgitation of stomach contents (gastroesophageal reflux) may be addressed by the addition of thickening agents to food, antacids, and maintaining an upright position after eating7.
Endocrinological Problems
Endocrinological issues require regular monitoring and may addressed using standard treatments. Some treatments that are used include levothyroxine for hypothyroidism, oral diazoxide for hyperinsulinemic hyperglycemia, and hormone replacement therapy for hypogonadism7.
Musculoskeletal Abnormalties
Musculoskeletal abnormalities can be managed by an orthopedist using standard therapies, which may include surgery. Calcium, vitamin D supplementation, and bisphosphonate have been used to treat low bone density and osteoporosisy7.
Therapies
Currently, there are no approved treatments for PMM2-CDG, but several therapies have been explored and two drugs are currently in human clinical trials.
Dietary Supplementation
Some studies have explored whether supplementing various sugars (dietary supplementation) into the diet of PMM2-CDG patients could improve overall protein glycosylation and potentially alleviate symptoms.
Galactose Supplementation
Oral supplementation of galactose has been explored as a treatment for PMM2-CDG, as this has helped treat some other CDG through various mechanisms. In a group of PMM2-CDG patients, increased consumption of galactose every day over 18 weeks did not result in statistically significant improvement of PMM2-symptoms but may have helped patients with mild symptoms more than those with severe disease46. Further studies are required to determine the effect of galactose supplementation in milder PMM2-CDG patients.
Mannose Supplementation
Several studies have explored the potential of mannose supplementation in improving the symptoms of PMM2-CDG by partially correcting altered mannose metabolism. Although mannose supplementation seemed promising in some model systems, oral supplementation of mannose in human PMM2-CDG patients did not lead to significant clinical improvement, despite some improvement in protein N-glycosylation13–15,47–51. Future studies on mannose supplementation are controversial due to the lack of perceived benefit to PMM2-patients.
Aldose Reductase Inhibitors
Epalrestat was approved for use in Japan three decades ago to treat diabetic neuropathy, a complication of diabetes, and is currently being explored for the treatment of PMM2-CDG17,42. It was initially identified in a drug repurposing screen, wherein known drugs were tested for their ability to restore PMM2 activity in patient-derived fibroblasts. Epalrestat is an aldose reductase inhibitor that works by preventing the conversion of glucose (elevated in diabetes) to sorbitol, which can otherwise accumulate inside cells and cause damage to peripheral neurons. The action of Epalrestat also results in an increase in glucose-1,6-bisphosphate, which helps stabilize and activate PMM2. By restoring enzyme activity, Epalrestat can treat the root cause of disease in some PMM2-CDG patients. Recently, promising results were seen in a child PMM2-CDG patient given oral epalrestat for one year, with significant improvement in growth and coordination and normalization of urine sorbitol levels42. As well, a phase 2 clinical trial is in progress (see below). Another aldose reductase inhibitor, AT-007 from Applied Therapeutics, is also being explored as a treatment for PMM2-CDG.
Mannose-1-Phosphate Replacement Therapy
Since PMM2 insufficiency results in lower levels of mannose-1-phosphate, with downstream effects on N-glycosylation, the direct supplementation of mannose-1-phosphate to PMM2-CDG patients has been explored to increase mannose-1-phospate concentrations in cells. Normally, mannose-1-phosphate does not enter cells as it is unable to pass through their cell membrane. However, Glycomine has developed a potential therapeutic, GLM101, that is designed to deliver mannose-1-phosphate directly into cells, potentially bypassing the PMM2 enzyme entirely.
Acetazolamide
Acetazolamide is currently being explored as a treatment for the movement and coordination issues associated with PMM2-CDG. Normally, acetazolamide is used as a diuretic medication to treat glaucoma and heart failure. It works by inhibiting carbonic anhydrase, which lowers blood pH and results in diuresis. This change in blood pH also affects the function of a calcium channel that is involved in nerve signalling, and results in improvement of symptoms in some disorders with cerebellar dysfunction16. In a study with 24 PMM2-CDG patients, acetazolamide treatment was well tolerated and did not result in any adverse events. Importantly, it resulted in improvement in the ability to repeat syllables and make coordinated movements in 20 of the patients, most within six weeks of treatment; patients with more severe clinical presentations and epilepsy showed a more substantial benefit from the treatment. An improvement of some coagulation parameters was also noted during treatment, but the cause of this is unknown. A larger clinical trial on the use of acetazolamide to treat the cerebellar manifestations of PMM2-CDG is in progress (see below).
Research Models
Several PMM2 research models have been generated including yeast, fruit fly, worm, zebrafish, mouse, human induced pluripotent stem cells (iPSC), patient-derived fibroblast models.
Yeast (S. cerevisiae)
SEC53 is orthologous to PMM2 in S. cerevisiae52. Mutations in SEC53 analogous to mutations in PMM2 were expressed in haploid, homozygous and heterozygous diploid yeast cells. Five mutations were explored in SEC53, F126L, E146K, R148H, V238M, and E100K corresponding to F119L, E139K, R141H, V231M, and E93A in PMM2, respectively. The effective of these mutations on yeast growth rate were measured, and it was found that residual enzyme activity was positively correlated with growth rate in both haploid and diploid cell lines. Consistent with other models, the R141H mutation was incompatible with growth.
Fly (D. melanogaster)
Pmm2-null mutant
A pmm2 null mutant was prepared using CRISPR/Cas9, which results in early lethality, psychomotor retardation, severely disrupted protein glycosylation, and impairment of the matrix metalloproteinase (MMP) pathway53.
Pmm2 knockdown
RNAi knockdown of neuronal PMM2 in flies results in severe ataxia, inability to fly and progressive loss of coordination, and later lethality53.
Nematode (C. elegans)
Pmm-2 homozygous null
Nematode PMM-2 is orthologous to human PMM2, and a large deletion in the pmm-2 gene (pmm-2 null) is larval lethal17.
Pmm-2 heterozygous null
Nematodes containing a pmm-2 null gene and a normal pmm-2 gene displayed normal growth and development17. Analysis of mRNA expression demonstrates that the mutant compensates for the null allele by overexpressing the genes at a transcriptional level, resulting in normal levels of PMM-2 formed from the normal allele.
Homozygous pmm-2F125L/F125L
The F125L mutation in nematode PMM-2 is analogous to the human F119L mutation, which is associated with a defect in PMM2 dimerization17. The homozygous nematode F125L genotype was prepared using CRISPR/Cas9 and is not larval lethal, nor does it result in growth or locomotor defects. However, it displays increased sensitivity to bortezomib (arrested as small larvae with morphological defects, or dead compared to viable wildtype), indicating an increased sensitivity to disruption of proteasomal processes and induction of ER stress.
Frog (X. laevis)
Pmm2 knockdown
In frogs, Pmm2 is orthologous to human PMM2. Pmm2 knockdown frog embryos can survive further into development than most other vertebrate model organisms, thereby facilitating the study of the role Pmm2 plays in cellular signalling and early development54. Pmm2 morpholino knockdown frogs display developmental delay and malformations in several major systems (eye, gut, heart, head, notochord, and stomach). Pmm2 knockdown frogs also display protein hypoglycosylation, including hypoglycosylation of Wnt5a. This results in impairment of the non-canonical Wnt signalling pathway, which plays an important role in early embryogenesis.
Fish (D. rerio)
Pmm2 morpholino knockdown
In zebrafish, Pmm2 is orthologous to human PMM2, and a morpholino knockdown model of PMM2-CDG was developed21. Morphant embryos had reduced PMM2 enzymatic activity, decreased LLO levels and N-linked glycosylation, and accumulation of mannose 6-phosphate. They displayed craniofacial cartilage defects and impaired motility associated with altered motor neurogenesis within the spinal cord, reminiscent of human PMM2-CDG.
Homozygous pmm2sa10150 mutants
A stable genetic PMM2-CDG zebrafish model was created, with zebrafish homozygous for a mutant pmm2 gene (pmm2sa10150) displaying pmm2 enzymatic activity at 20% of wildtype20. Embryos homozygous for the pmm2sa10150 allele exhibited craniofacial cartilage defects, loss of movement starting at 5-6 days post fertilization, altered motor neurogenesis, and death by 13-14 days post fertilization.
Mouse (M. musculus)
Pmm2-/- Constitutive Knockout
Murine Pmm2, produced by the Pmm2 gene, is orthologous to human PMM2. The whole body knockout is incompatible with life, causing embryonic lethality by 3.5 days post coitum due to either failure in embryonic implantation or death prior to implantation50.
Homozygous Pmm2R137H/R137H
This genotype is analogous to the human homozygous R141H mutation and is embryonic lethal in mice by 5.5 days post coitum50. This lethality is consistent with the absence of humans reported to be homozygous for the R141H mutation.
Homozygous Pmm2F118L/F118L
The F118L mutation is analogous to the F122L mutation in human PMM2, which is an artificial mutation predicted to cause very mild loss in enzyme activity. Mice homozygous with this mutation are viable with no phenotype50.
Heterozygous Pmm2R137H/F118L
The R137H/F118L genotype results in stunted embryonic growth and lethality between 9.5-10.5 days post coitum50. Oral mannose supplementation to pregnant dams rescues these embryos, allowing them to survive paste weaning with no significant differences in development or glycosylation levels. Pups that survived weaning demonstrated normal phenotypes even after removal of mannose supplementation, which demonstrates the importance of Pmm2 function in embryonic development.
Heterozygous Pmm2R137H/F115L
This genotype corresponds to the most common human PMM2-CDG genotype (R141H/P119L), and often resulted in prenatal lethality after 12.5 days post coitum that was not reversed by mannose supplementation to the dams55. Surviving mice demonstrated delayed growth and impaired protein glycosylation, with approximately half dying within 65 days after birth.
Homozygous Pmm2 F115L/F115L
This genotype causes significant embryonic lethality which could be reversed upon feeding the dams mannose before and during pregnancy55.
Human Cell Lines
Patient derived fibroblasts
Mannose supplementation in patient derived fibroblasts results in partially corrected synthesis of the LLO, and partially restores abnormally low intracellular levels of GDP-mannose49. Patient derived fibroblasts have also been used to assay compounds for the ability to act as pharmacological chaperones for PMM256.
Induced pluripotent stem cells (iPSCs)
Fibroblasts from an R141H/F119L PMM2-CDG patient were reprogrammed into pluripotent stem cells using lentiviral manipulation57. These PMM2-deficient induced pluripotent stem cells (iPSCs) displayed reduced N-glycan levels but were able to be differentiated into hepatic, neural progenitor, and cardiomyogenic cells carrying the faulty PMM2 alleles. The hypoglycosylation in the induced pluripotent stem cells could be rescued by adding mannose to the culture media.
Clinical Studies
Active
Acetazolamide Efficacy in Ataxia in PMM2-CDG (NCT04679389)
The Mayo Clinic, in collaboration with Seattle Children’s Hospital and the Children’s Hospital of Philadelphia, is conducting a Phase 2/Phase 3 on the use of acetazolamide to improve the neurological symptoms associated with PMM2-CDG (particularly ataxia) in patients four years and older. A secondary objective is to monitor the effect of acetazolamide on biomarkers associated with PMM2-CDG.
Natural History Study Protocol in PMM2-CDG (CDG-Ia) (NCT03173300)
Glycomine, Inc. is conducting a 4-year natural history study of on patients with PMM2-CDG. The goal of this study is to study the signs and symptoms of PMM2-CDG and how they change over time.
Oral Epalrestat Therapy in Pediatric Subjects with PMM2-CDG (NCT04925960)
The Mayo Clinic is conducting Phase 2 clinical trial on the use of oral Epalrestat therapy in pediatric PMM2-CDG patients. The purpose of the study is to evaluate the safety, tolerability, metabolic improvement and likely efficacy of Epalrestat at treating PMM2-CDG.
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including PMM2-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Completed
Sorbitol Is a Severity Biomarker for PMM2-CDG with Therapeutic Implications
The Mayo Clinic, in collaboration with Maggie’s Pearl LLC, conducted a single patient compassionate use study on the effect of oral epalrestat in a child with PMM2-CDG for a one year period. No adverse events were reported, and the patient experienced significantly improved growth, improved coordination, and normalization of urinary sorbitol levels and blood transferrin glycosylation.
Using D-Galactose as a Food Supplement in Congenital Disorders of Glycosylation (NCT02955264)
Tulane University conducted an open-label pilot trial which explored D-galactose supplementation in nine patients with PMM2-CDG. Overall, there was no significant improvement in patients. Some patients with mild disease displayed some positive changes, but larger placebo controlled trials are necessary to determine the suitability of D-galactose administration as a supportive therapy in PMM2-CDG46.
AZATAX Acetazolamide safety and efficacy in cerebellar syndrome in PMM2 congenital disorder of glycosylation (PMM2-CDG)
The CDG Spanish Consortium evaluated the effect of acetazolamide on PMM2-CDG patients in a Phase I/II clinical trial. Acetazolamide was well tolerated and improved the cerebellar syndrome (e.g. ataxia) present in most PMM2-CDG patients, but the long-term effects on kidney function should be addressed in future studies.
Organizations/Groups
Maggie’s Pearl LLC
Amour Science Foundation
Publications
PMM2-CDG Scientific Articles on PubMed
International Clinical Guidelines for the Management of PMM2-CDG
Additional Resources
PMM2-CDG Clinical Utility Gene Card
PMM2-CDG Infographics
IEMbase
GeneReviews
OMIM
OrphaNet
NORD
GARD
MedlinePlus
Genetic Testing Registry
ClinVar
NIH
GeneCards
UniProt
References
- Matthijs, G. et al. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome). Nat. Genet. 16, 88–92 (1997).
- Jaeken, J., Lefeber, D. & Matthijs, G. Clinical utility gene card for: Phosphomannomutase 2 deficiency. Eur. J. Hum. Genet. 2014 228 22, 1054–1054 (2014).
- Jaeken, J. et al. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin. Chim. Acta 144, 245–247 (1984).
- Jaeken, J. et al. Familial psychomotor retardation with markedly fluctuating serum prolactin, FSH and GH levels, partial TBG-deficiency, increased serum arylsulphatase A and increased CSF protein: a new syndrome?: 90. Pediatr. Res. 1980 142 14, 179–179 (1980).
- Van Schaftingen, E. & Jaeken, J. Phosphomannomutase deficiency is a cause of carbohydrate-deficient glycoprotein syndrome type I. FEBS Lett. 377, 318–320 (1995).
- Gámez, A., Serrano, M., Gallego, D., Vilas, A. & Pérez, B. New and potential strategies for the treatment of PMM2-CDG. Biochim. Biophys. Acta - Gen. Subj. 1864, 129686 (2020).
- Altassan, R. et al. International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: Diagnosis, treatment and follow up. J. Inherit. Metab. Dis. 42, 5–28 (2019).
- Matthijs, G. et al. Mutations in PMM2 That Cause Congenital Disorders of Glycosylation, Type Ia (CDG-Ia). Hum. Mutat. 16, 386–394 (2000).
- Schollen, E., Kjaergaard, S., Legius, E. Schwartz, M. & Matthijs, G. Lack of Hardy-Weinberg equilibrium for the most prevalent PMM2 mutation in CDG-Ia (congenital disorders of glycosylation type Ia). Eur. J. Hum. Genet. 8, 367–371 (2000).
- Yildiz, Y. et al. Genotypes and estimated prevalence of phosphomannomutase 2 deficiency in Turkey differ significantly from those in Europe. Am. J. Med. Genet. Part A 182, 705–712 (2020).
- Vals, M.A., Pajusalu, S., Kals, M., Mägi, R. & Õunap, K. The Prevalence of PMM2-CDG in Estonia Based on Population Carrier Frequencies and Diagnosed Patients. in JIMD Reports, Volume 39 (eds. Morava, E. et al.) 13–17 (Springer Berlin Heidelberg, 2018). doi:10.1007/8904_2017_41.
- Lam, C. & Krasnewich, D. M. PMM2-CDG. in GeneReviews [Internet] (eds. Adam, M. P., Ardinger, H. H., Pagon, R. A. & Wallace, S. E.) (University of Washington, 2005).
- Witters, P. et al. Spontaneous improvement of carbohydrate-deficient transferrin in PMM2-CDG without mannose observed in CDG natural history study. Orphanet J. Rare Dis. 2021 16, 1–5 (2021).
- Taday, R., Grüneberg, M., DuChesne, I., Reunert, J. & Marquardt, T. Dietary mannose supplementation in phosphomannomutase 2 deficiency (PMM2-CDG). Orphanet J. Rare Dis. 15, 1–8 (2020).
- Taday, R. et al. Mannose supplementation in PMM2-CDG. Orphanet J. Rare Dis. 16, 1–3 (2021).
- Martínez-Monseny, A.F. et al. AZATAX: Acetazolamide safety and efficacy in cerebellar syndrome in PMM2 congenital disorder of glycosylation (PMM2-CDG). Ann. Neurol. 85, 740–751 (2019).
- Iyer, S. et al. Repurposing the aldose reductase inhibitor and diabetic neuropathy drug epalrestat for the congenital disorder of glycosylation PMM2-CDG. Dis. Model. Mech. 12, (2019).
- Vaes, L. et al. PMM2-CDG caused by uniparental disomy: Case report and literature review. J. Inherit. Metab. Dis. 54, 16–21 (2020).
- Pérez, B. et al. Segmental uniparental disomy leading to homozygosity for a pathogenic mutation in three recessive metabolic diseases. Mol. Genet. Metab. 105, 270–271 (2012).
- Klaver, E. J. et al. Protease-dependent defects in N-cadherin processing drive PMM2-CDG pathogenesis. JCI insight 6, (2021).
- Cline, A. et al. A zebrafish model of PMM2-CDG reveals altered neurogenesis and a substrate-accumulation mechanism for N-linked glycosylation deficiency. Mol. Biol. Cell 23, 4175–4187 (2012).
- Gao, N. et al. Mannose-6-phosphate regulates destruction of lipid-linked oligosaccharides. Mol. Biol. Cell 22, 2994–3009 (2011).
- Schollen, E. et al. Characterization of two unusual truncating PMM2 mutations in two CDG-Ia patients. Mol. Genet. Metab. 90, 408–413 (2007).
- Vuillaumier-Barrot, S. et al. PMM2 intronic branch-site mutations in CDG-Ia. Mol. Genet. Metab. 87, 337–340 (2006).
- Kjaergaard, S., Skovby, F. & Schwartz, M. Absence of homozygosity for predominant mutations in PMM2 in Danish patients with carbohydrate-deficient glycoprotein syndrome type 1. Eur. J. Hum. Genet. 6, 331–336 (1998).
- Matthijs, G., Schollen, E., Van Schaftingen, E., Cassiman, J. J. & Jaeken, J. Lack of Homozygotes for the Most Frequent Disease Allele in Carbohydrate-Deficient Glycoprotein Syndrome Type 1A. Am. J. Hum. Genet. 62, 542–550 (1998).
- Lonlay, P. de et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J. Med. Genet. 38, 14–19 (2001).
- Barone, R. et al. A nationwide survey of PMM2-CDG in Italy: high frequency of a mild neurological variant associated with the L32R mutation. J. Neurol. 2014 2621 262, 154–164 (2014).
- Tayebi, N. et al. A deletion–insertion mutation in the phosphomannomutase 2 gene in an African American patient with congenital disorders of glycosylation-Ia. Am. J. Med. Genet. 108, 241–246 (2002).
- Matthijs, G., Schollen, E., Heykants, L. & Grünewald, S. Phosphomannomutase Deficiency: The Molecular Basis of the Classical Jaeken Syndrome (CDGS Type Ia). Mol. Genet. Metab. 68, 220–226 (1999).
- Vals, M.A. et al. Three families with mild PMM2-CDG and normal cognitive development. Am. J. Med. Genet. Part A 173, 1620–1624 (2017).
- Vaes, L. et al. Genotype-Phenotype Correlations in PMM2-CDG. Genes (Basel). 12, (2021).
- Cabezas, O. R. et al. Polycystic Kidney Disease with Hyperinsulinemic Hypoglycemia Caused by a Promoter Mutation in Phosphomannomutase 2. J. Am. Soc. Nephrol. 28, 2529–2539 (2017).
- Vuillaumier-Barrot, S. et al. Expanding the Spectrum of PMM2-CDG Phenotype. in JIMD Reports - Case and Research Reports, 2012/2 123–125 (Springer Berlin Heidelberg, 2012). doi:10.1007/8904_2011_114.
- Francisco, R. et al. The challenge of CDG diagnosis. Mol. Genet. Metab. 126, 1–5 (2019).
- Grünewald, S., Schollen, E., Van Schaftingen, E., Jaeken, J. & Matthijs, G. High Residual Activity of PMM2 in Patients’ Fibroblasts: Possible Pitfall in the Diagnosis of CDG-Ia (Phosphomannomutase Deficiency). Am. J. Hum. Genet. 68, 347–354 (2001).
- Patterson, M. C. Metabolic mimics: The disorders of N-linked glycosylation. Semin. Pediatr. Neurol. 12, 144–151 (2005).
- Hahn, S. H., Minnich, S. J. & O’brien, J. F. Stabilization of hypoglycosylation in a patient with congenital disorder of glycosylation type Ia. J Inherit Metab Dis 29, 235–237 (2006).
- Al Teneiji, A. et al. Phenotypic and genotypic spectrum of congenital disorders of glycosylation type I and type II. Mol. Genet. Metab. 120, 235–242 (2017).
- Sturiale, L., Barone, R. & Garozzo, D. The impact of mass spectrometry in the diagnosis of congenital disorders of glycosylation. J. Inherit. Metab. Dis. 34, 891–899 (2011).
- Van Scherpenzeel, M., Steenbergen, G., Morava, E., Wevers, R. A. & Lefeber, D. J. High-resolution mass spectrometry glycoprofiling of intact transferrin for diagnosis and subtype identification in the congenital disorders of glycosylation. Transl. Res. 166, 639-649.e1 (2015).
- Ligezka, A. N. et al. Sorbitol Is a Severity Biomarker for PMM2-CDG with Therapeutic Implications. Ann. Neurol. 90, 887–900 (2021).
- Feldman, B. J. & Rosenthal, D. Carbohydrate-Deficient Glycoprotein Syndrome-Associated Pericardial Effusion Treated with Corticosteroids and Salicylic Acid. Pediatr. Cardiol. 23, 469–471 (2002).
- Monagle, P. Diagnosis and Management of Deep Venous Thrombosis and Pulmonary Embolism in Neonates and Children. Semin. Thromb. Hemost. 38, 683–690 (2012).
- Kearon, C. et al. Antithrombotic Therapy for VTE Disease: CHEST Guideline and Expert Panel Report. Chest 149, 315–352 (2016).
- Witters, P. et al. D-galactose supplementation in individuals with PMM2-CDG: results of a multicenter, open label, prospective pilot clinical trial. Orphanet J. Rare Dis. 16, (2021).
- Mayatepek, E. & Kohlmuller, D. Mannose supplementation in carbohydrate-deficient glycoprotein syndrome type I and phosphomannomutase deficiency. Eur. J. Pediatr. 157, 605–612 (1998).
- Kjaergaard, S. et al. Failure of short-term mannose therapy of patients with carbohydrate-deficient glycoprotein syndrome type 1A. Acta Pædiatrica 87, 884–888 (1998).
- Rush, J.S., Panneerselvam, K., Waechter, C.J. & Freeze, H.H. Mannose supplementation corrects GDP-mannose deficiency in cultured fibroblasts from some patients with Congenital Disorders of Glycosylation (CDG). Glycobiology 10, 829–835 (2000).
- Schneider, A. et al. Successful prenatal mannose treatment for congenital disorder of glycosylation-Ia in mice. Nat. Med. 2011 181 18, 71–73 (2011).
- Panneerselvam, K. & Freeze, H.H. Mannose corrects altered N-glycosylation in carbohydrate-deficient glycoprotein syndrome fibroblasts. J. Clin. Invest. 97, 1478–1487 (1996).
- Lao, J. P. et al. Yeast Models of Phosphomannomutase 2 Deficiency, a Congenital Disorder of Glycosylation. G3 Genes|Genomes|Genetics 9, 413–423 (2019).
- Parkinson, W. M. et al. Synaptic roles for phosphomannomutase type 2 in a new Drosophila congenital disorder of glycosylation disease model. Dis. Model. Mech. 9, 513–527 (2016).
- Himmelreich, N., Kaufmann, L. T., Steinbeisser, H., Körner, C. & Thiel, C. Lack of phosphomannomutase 2 affects Xenopus laevis morphogenesis and the non-canonical Wnt5a/Ror2 signalling. J. Inherit. Metab. Dis. 2015 386 38, 1137–1146 (2015).
- Chan, B. et al. A mouse model of a human congenital disorder of glycosylation caused by loss of PMM2. Hum. Mol. Genet. 25, 2182–2193 (2016).
- Yuste-Checa, P. et al. Pharmacological Chaperoning: A Potential Treatment for PMM2-CDG. Hum. Mutat. 38, 160–168 (2017).
- Thiesler, C. T. et al. Glycomic Characterization of Induced Pluripotent Stem Cells Derived from a Patient Suffering from Phosphomannomutase 2 Congenital Disorder of Glycosylation (PMM2-CDG) *. Mol. Cell. Proteomics 15, 1435–1452 (2016).