
Lay Summary
ALG11-CDG, formerly known as CDG-Ip, is a rare inherited condition that affects many parts of the body. To date, more than 15 patients have reported in the literature. ALG11-CDG is classified as a disorder of N-linked protein glycosylation. ALG11-CDG is caused by mutations in both copies of the ALG11 gene which provides instructions for making an enzyme that attaches the simple sugar mannose to growing sugar chains during N-glycosylation. Mutations in the ALG11 gene cause proteins to have incomplete or absent sugar chains. Symptoms of ALG11-CDG begin in infancy and are primarily characterized by developmental delay, epilepsy, abnormal facial features, gastrointestinal and vision problems. Several screening tests are available for ALG11-CDG, but a definitive diagnosis is achieved through genetic testing. Treatment is focused on the management of specific symptoms and preventing complications.
Overview
ALG11-CDG is a rare autosomal recessive genetic disorder. The ALG11 (asparagine-linked glycosylation 11 homolog) gene encodes a mannosyltransferase enzyme that catalyzes the attachment of the fourth and fifth of nine mannose residues during synthesis of the lipid linked oligosaccharide (LLO) in the cytosol of the endoplasmic reticulum (ER). The synthesis of the LLO is a precursor step to N-glycosylation. Deficiency of the ALG11 enzyme results in the incomplete assembly of the LLO, leading to insufficient N-glycosylation of glycoproteins.
The first reported case of ALG11-CDG was in 20101 and more than 15 patients have reported in the literature to date1–9 . Symptoms begin in infancy, presenting as neurological developmental delay, epilepsy, dysmorphic features, gastrointestinal and vision problems7. A diagnosis can be determined through transferrin analysis, LLO analysis, and analysis of a unique biomarker, GP130, although a definitive diagnosis can only be achieved through molecular genetic testing. There are currently no approved treatments for ALG11-CDG7.
Synonyms
- CDG-Ip
- Congenital disorder of glycosylation type 1p
- Mannosyltransferase 4-5 deficiency
Inheritance
ALG11-CDG is an autosomal recessive disorder meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The ALG11 gene encodes a mannosyltransferase enzyme (ALG11)7. Mannosyltransferases are enzymes that enable the transfer of mannose during glycosylation. ALG11 is located in the ER membrane, facing the cytosol, where it has a role in the assembly of the LLO, a precursor for protein N-glycosylation.
The ALG11 enzyme sequentially transfers the fourth and fifth mannose residues to the growing oligosaccharide prior to its attachment to a protein7. Mutations in the ALG11 gene lead to the production of an enzyme with reduced or no activity and an accumulation of the incomplete LLO (Dol-PP-GlcNAc2Man3) causing proteins to be insufficiently glycosylated (hypoglycosylation)7,10.
LLO synthesis
N-glycosylation is the process by which an oligosaccharide is attached to the nitrogen atom of asparagine residues on proteins. N-glycosylation is initiated in the ER and begins with the synthesis of an LLO11. The LLO is comprised of a 14-sugar oligosaccharide attached to the lipid carrier dolichol pyrophosphate (Dol-PP). The 14-sugar residues are 2 N-acetylglucosamine, 9 mannose, and 3 glucose residues. Once assembled, the oligosaccharide is transferred en bloc to proteins and undergoes further processing in the ER and Golgi11. Once the oligosaccharide is attached to a protein, it is referred to as an N-glycan.
LLO synthesis is carried out by a series of enzymes encoded by the ALG genes and can be divided into two phases: Phase I and Phase II 11 (Figure 1).
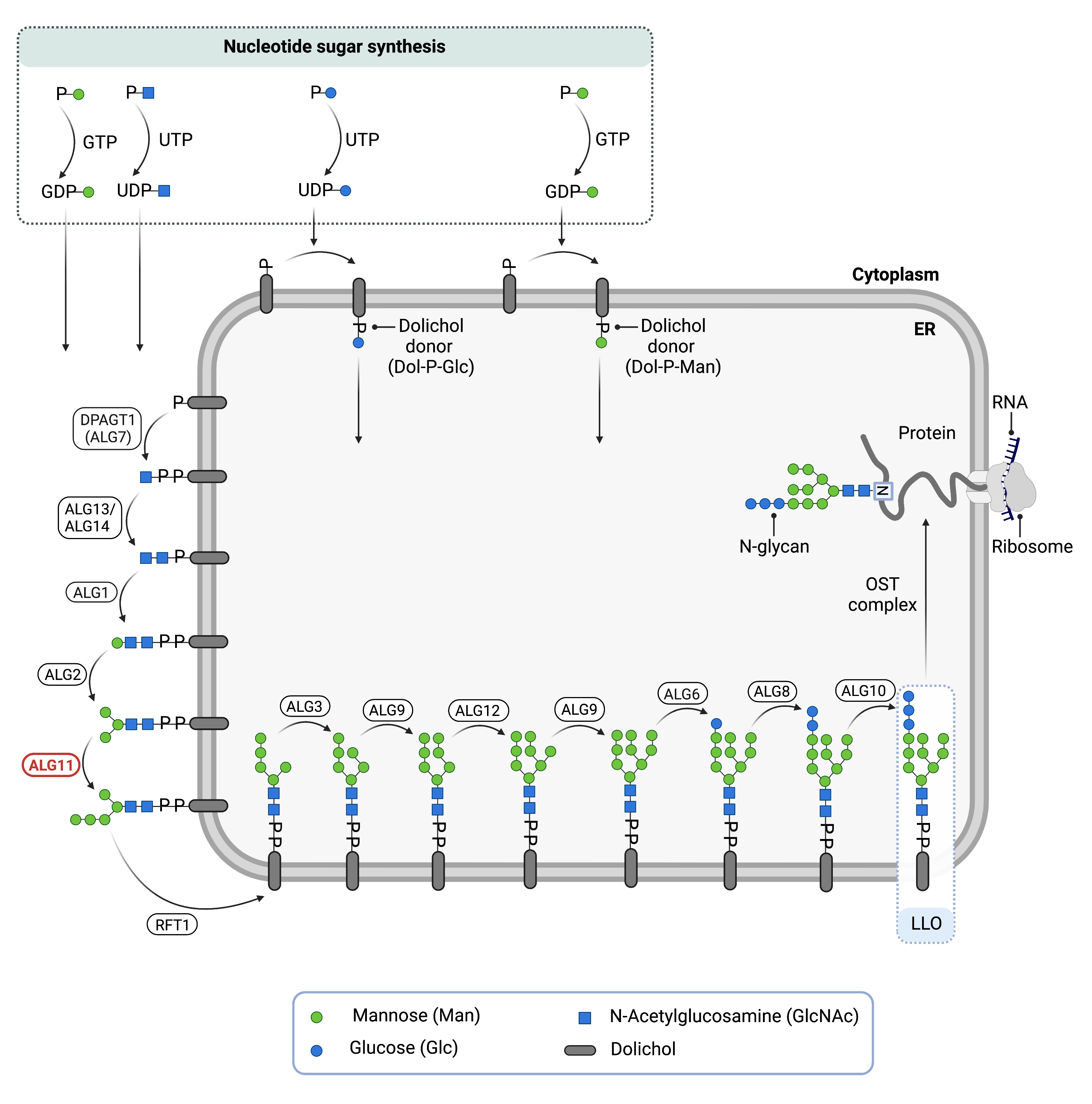
Figure 1. Role of ALG11 in glycosylation.
ALG11 is an enzyme involved in synthesizing the lipid-linked oligosaccharide (LLO) in N-glycosylation. ALG11 catalyzes the transfer of fourth and fifth mannose residues from the nucleotide sugar, GDP-mannose, onto the growing LLO on the cytosolic side of endoplasmic the reticulum membrane.
Phase I
Phase I of LLO synthesis takes place on the cytoplasmic side of the ER. It begins with the sequential attachment of two N-Acetylglucosamine (GlcNAc2) and five mannose (Man5) residues to Dol-PP by several ER enzymes. GlcNAc and mannose are transferred from nucleotide sugars UDP-GlcNAc and GDP-mannose, respectively. ALG11 catalyzes the transfer of the fourth and fifth mannose resides from GDP-mannose onto the growing LLO. The intermediate structure, Dol-PP-GlcNAc2Man5 is translocated across the ER membrane into the lumen via the RFT1 enzyme11,12.
Phase II
Phase II of LLO synthesis takes place in the ER lumen. Once in the lumen, four annose residues followed by three glucose (Glc3) residues are added to the intermediate structure, generating the complete LLO, Dol-PP-GlcNAc2Man9Glc3. Mannose and Glucose are transferred from glycosyl donors Dol-P-mannose and Dol-P-glucose, which are formed on the cytoplasmic side of the ER and must also be flipped across the ER membrane11.
Once assembled, the oligosaccharide is transferred en bloc from Dol-PP to asparagine residues of newly synthesized protein via the enzyme oligosaccharyltransferase (OST), resulting in N-glycosylation of the protein11. The activity of OST is highly specific for the completely assembled 14-sugar oligosaccharide, GlcNAc2Man9Glc3.
In the absence of ALG11, the LLO is missing 6 mannose residues and 3 glucose residues, and therefore is incompletely assembled. This results in reduced transfer efficiency of the oligosaccharide by OST, causing an accumulation of the incomplete LLO and hypoglycosylation of glycoproteins which lack N-glycans.
Disease Mechanism
Mutations in the ALG11 gene lead to the production of an abnormal enzyme with reduced or no activity. In the absence of ALG11, the LLO is incompletely assembled and accumulates inside the ER after being transferred by OST10.
Mutations
The ALG11 gene is located on chromosome 13 (13q14.3). 10 pathogenic variants in ALG11 have been reported in the literature to date. The most common variants are either homozygous or compound heterozygous for point mutations or insertion-deletions (indels). One patient reported with a missense mutation in trans to a whole gene deletion appears to have a less severe phenotype compared to other reported mutations7.
Signs & Symptoms
Clinical Presentation
Individuals with ALG11-CDG develop signs and symptoms that may be detected in infancy, as early as a few days old. ALG11-CDG is primarily characterized by developmental disability, epilepsy, visual problems, microcephaly, hypotonia and dysmorphic features 6,7:
- Neurological – Developmental disability, epilepsy, decreased muscle tone (hypotonia), smaller than average head at birth (microcephaly), increased muscle tone (peripheral hypertonia), impaired hearing and overactive or over-responsive reflexes (hyperreflexia).
- Ophthalmological: strabismus, poor visual tracking
- Dysmorphic features – long philtrum, abnormal jaw structure
- Gastrointestinal abnormalities – poor feeding and recurrent vomiting.
Other less common symptoms may include oscillations of body temperature, ataxia, papillary atrophy, retinal dystrophy, convergent strabismus, cerebral atrophy, and white matter abnormalities.
Biochemical Abnormalities
There have been no reports of significant biochemical abnormalities in ALG11-CDG patients6,7.
Classification
ALG11-CDG is classified as an N-linked glycosylation disorder.
Under the former CDG classification system, ALG11-CDG is classified as a Type 1 CDG, which arise due to defects in the synthesis of oligosaccharides or their transfer to proteins.
Diagnosis
Although a diagnosis of ALG11-CDG may be suspected based on presentation of symptoms and a detailed patient history, direct molecular genetic testing is the only definitive diagnostic test. Screening in suspected patients begins with a blood test to analyze serum transferrin. LLO analysis in patient fibroblasts may also be carried out following transferrin screening7. Additionally, protein GP130 has been identified as a potential biomarker for ALG11-CDG and has been shown to be hypoglycosylated.
Transferrin Analysis
Individuals with ALG11-CDG show a type 1 pattern by transferrin isoelectric focusing (TIEF) or mass spec analysis of transferrin. Type 1 patterns are observed in CDG that arise due to defects in LLO synthesis and are characterized by a decrease in tetrasialo-transferring and an increase in di-sialo and a-sialo transferrin glycoforms.
LLO Analysis
LLO analysis of ALG11-CDG fibroblasts show an increase in the intermediate N-glycan structures, GlcNAc2Man3 and GlcNAc2Man4 , which are lacking 6 mannose residues and 3 glucose residues7.
Biomarkers
The protein GP130 was found to be hypoglycosylated in patient fibroblasts from two ALG11-CDG patients; abnormally truncated GP130 isoforms was also reported7,9.
Prognosis
The prognosis of ALG11-CDG may vary depending on severity of an individual’s symptoms. The broad clinical spectrum may result in premature death in some patients. The oldest patient reported is 14 years old, with all other patients ranging from 2 months to 8 years1,6,7.
Management
Management of symptoms may include combinations of physical therapy, occupational therapy, speech or vision therapy and palliative measures.
Therapies
There are currently no treatment options available for ALG11-CDG. Treatment is focused on management of symptoms and prevention of complications.
Research Models
Several ALG11 research models have been generated including yeast, fly, and mouse models.
Yeast (S. cerevisiae)
Alg11 null mutants have been generated. Mutants are sensitive to elevated temperatures and to cell-wall affecting drugs such as caffeine and Calcofluor White. Mutants also have slowed growth. (YeastGenome).
Fly (D. melanogaster)
Drosophila gene Alg11 (ALG11, CG11306, FBgn0037108) is orthologous to ALG11. It is predicted to have mannosyltransferase activity and be involved in protein glycosylation. It is expressed in the adult drosophila head and is a potential model for human disease ALG11-CDG (FlyBase).
Mouse (M. musculus)
Several mouse models have been generated including a complete knockout mouse and conditional knockout embryonic stem cell lines.
Homozygous Alg11-/- and heterozygous Alg11-/- knockout mice
Alg11tm1.1(KOMP)Vlcg homozygous and heterozygous mutant mice have been generated with an exon deletion. Homozygous mice live from E9.5 to early adult and phenotypically present with embryonic lethality prior to organogenesis or preweaning lethality and complete penetrance. Heterozygous mutants live to early adulthood and phenotypically show a decreased grip strength (IMPC).
Alg11 knockout ES cell line
Embryonic Stem (ES) cell line Alg11tm1e(EUCOMM)Wtsi and Alg11tm1(KOMP)Vlcg are targeted knockout/null mutations of Alg11 (MGI, MGI).
Human Cell lines
Patient Derived Fibroblasts
ALG11-CDG patient fibroblasts were used to investigate pathogenicity of ALG11 mutation variants1,6,7. Additionally, abnormally truncated isoforms of the protein GP130 were identified in patient samples that were unaffected in control fibroblasts samples. This suggests that GP130 may be used as a biomarker for diagnosis of ALG11-CDG7.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including ALG11-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDGs, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Publications
ALG11-CDG Scientific Articles on PubMed
Additional Resources
References
- Rind, N. et al. A severe human metabolic disease caused by deficiency of the endoplasmatic mannosyltransferase hALG11 leads to congenital disorder of glycosylation-Ip. Human Molecular Genetics 19, (2010).
- Mulkey, S. B. et al. Arrest of Fetal Brain Development in ALG11-Congenital Disorder of Glycosylation. Pediatric Neurology 94, (2019).
- Thiel, C. et al. Improved diagnostics lead to identification of three new patients with congenital disorder of glycosylation-Ip. Human Mutation 33, (2012).
- Pereira, A. G. et al. Epileptic spasms in congenital disorders of glycosylation. Epileptic Disorders 19, (2017).
- al Teneiji, A. et al. Phenotypic and genotypic spectrum of congenital disorders of glycosylation type I and type II. Molecular Genetics and Metabolism 120, (2017).
- Regal, L. et al. ALG11-CDG: Three novel mutations and further characterization of the phenotype. Molecular Genetics and Metabolism Reports 2, (2015).
- Haanpää, M. K. et al. ALG11‐CDG syndrome: Expanding the phenotype. American Journal of Medical Genetics Part A 179, (2019).
- Silver, G. et al. Prevalence of Congenital Disorders of Glycosylation in Childhood Epilepsy and Effects of Anti-Epileptic Drugs on the Transferrin Isoelectric Focusing Test. Genes (Basel) 12, (2021).