
Lay Summary
Congenital disorders of glycosylation (CDG) arise from abnormalities in glycosylation – the process by which sugar chains (glycans) are attached to proteins or fats (lipids). Depending on how these molecules are modified with sugars, glycosylation can be classified into different pathways such as N-glycosylation, O-glycosylation, GPI anchor biosynthesis and lipid glycosylation. All of these glycosylation pathways require sugars and enzymes, as well as transportation systems to ensure these substances are delivered to the correct cellular location to participate in glycosylation. Consequently, disorders of multiple glycosylation pathways arise from defects in substances shared between two or more glycosylation pathways. These disorders have varied clinical features, and diagnosis often relies on screening for abnormal patterns of sugars on proteins and genetic testing1,2.
Overview
Glycosylation is a process where enzymes (glycosyltransferases) transfer simple sugars (monosaccharides) to molecules, such as proteins, lipids or other sugars. Glycosylation can be divided into different pathways such as N-glycosylation, O-glycosylation, glycosylphosphatidylinositol (GPI) anchor biosynthesis and lipid glycosylation3. All glycosylation pathways require high-energy forms of monosaccharides, called activated sugars, or enzymes to be able to transfer the monosaccharide to the protein, lipid or glycan. These activated sugars are typically monosaccharides linked to a group of molecules called nucleosides (forming nucleotide sugars) or linked to the lipid dolichol through high-energy phosphate bonds4,5. After the first monosaccharide is attached to the lipid, protein or sugar, additional monosaccharides can be sequentially transferred to the attached monosaccharide, generating sugar chains, called glycans. As glycosylation can occur in different locations in the cell, such as the cytosol, endoplasmic reticulum (ER) or Golgi complex (Golgi), transportation systems exist to deliver the different glycosylation components (such as enzymes and activated sugars) to the right location6,7.
Disorders of multiple glycosylation pathways arise from defects in components that are shared between different glycosylation pathways, which can include enzymes, sugars, and transportation systems8. These disorders of multiple glycosylation pathways can be subcategorised into4:
- Disorders of monosaccharide synthesis and interconversion
- Disorders of dolichol metabolism
- Disorders of nucleotide sugar synthesis and transport
- Disorders of vesicular trafficking
- Disorders of Golgi homeostasis
- Other disorders of multiple glycosylation pathways
Disorders of Monosaccharide Synthesis and Interconversion
Disorders of monosaccharide synthesis and interconversion arise from mutations in genes encoding enzymes involved in sugar metabolism. Monosaccharides can be imported into the cell, recycled from degraded glycans, or be prepared by altering the structure of existing monosaccharides to generate different sugars (interconversion) (Figure 1). Monosaccharide synthesis and interconversion relies on enzymes that can add and remove molecules, such as phosphate, to and from the monosaccharide, or rearrange position of atoms or the way they are put together in the monosaccharides9–12.
Disorders of monosaccharide synthesis and interconversion include:
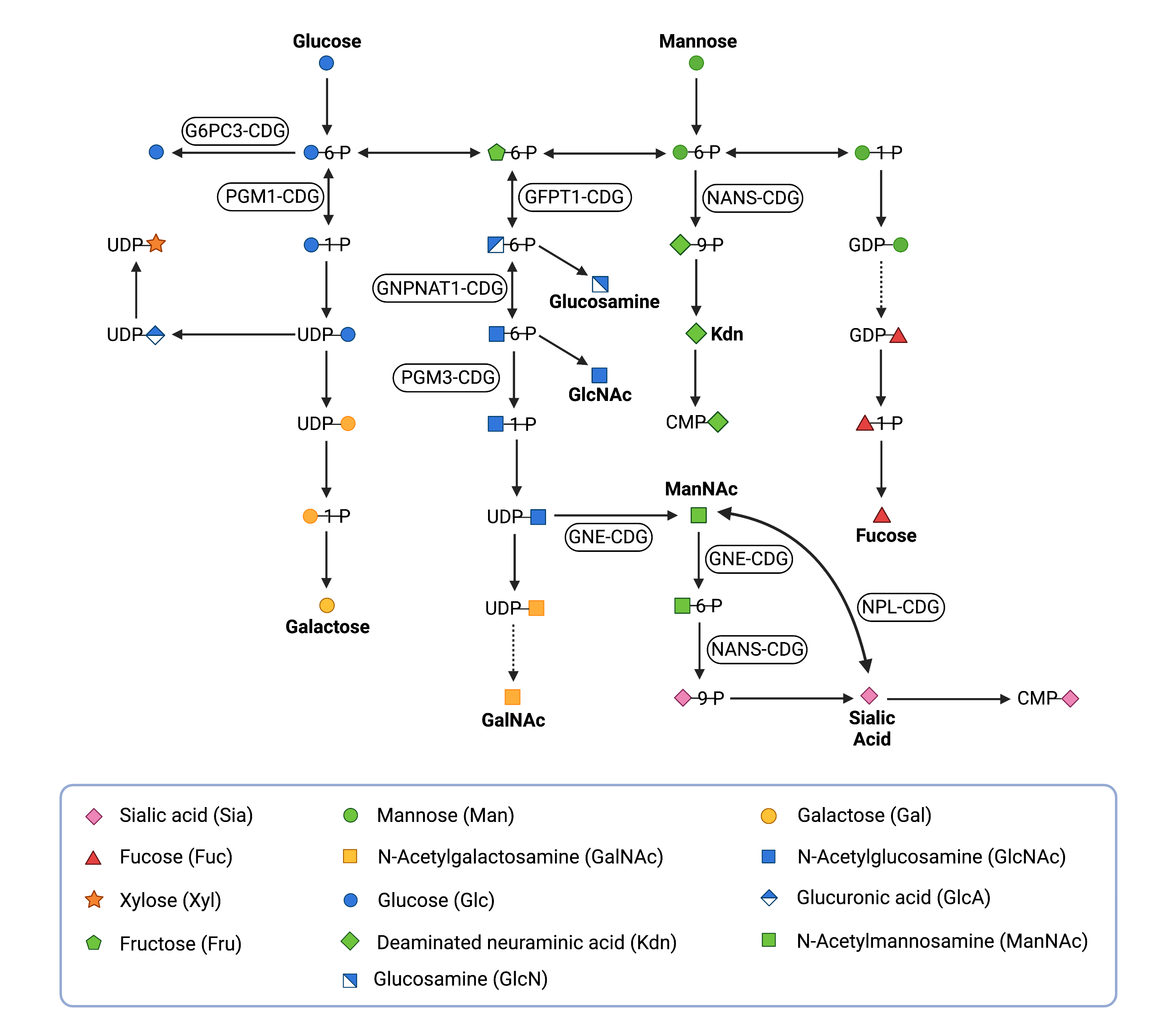
Figure 1. Overview of monosaccharide synthesis and interconversion, with associated CDG types.
Monosaccharides can be imported into the cell or recycled from glycans that have been broken down; a monosaccharide can be converted into another monosaccharide (monosaccharide interconversion).
Disorders of Dolichol Metabolism
Dolichol is a lipid that can be found in the membrane of the ER and is needed for a variety of glycosylation pathways13. Defects in dolichol metabolism can therefore lead to disorders that impair multiple glycosylation pathways14.
Cells produce and modify dolichol (Dol) through a series of enzymatic reactions (Figure 2). Dolichol is required for the formation of dolichol-linked activated sugars, and is thus critical for N-, O- and C-glycosylation and GPI anchor biosynthesis2,13,15. To generate these activated sugars, enzymes first modify the synthesised dolichol with a phosphate molecule (Dol-P) using enzymes called kinases15. Glycosyltransferases then attach monosaccharides (mannose or glucose) to Dol-P, generating the activated sugars Dol-P-mannose (Dol-P-Man) or Dol-P-glucose (Dol-P-Glc), respectively. Glycosyltransferases can transfer monosaccharides from these dolichol-based activated sugars to proteins, lipids or other glycans. Additionally, Dol-P acts as a lipid carrier of oligosaccharides in the early stages of N-glycosylation. Here, glycosyltransferases sequentially transfer activated monosaccharides from nucleotide sugars or dolichol-linked sugars to Dol-P to generate a lipid-linked oligosaccharide (LLO)12. Upon transfer of the monosaccharide or oligosaccharide from Dol-P and Dol-PP, dolichol is released and re-phosphorylated14,15.
Disorders of dolichol metabolism include:
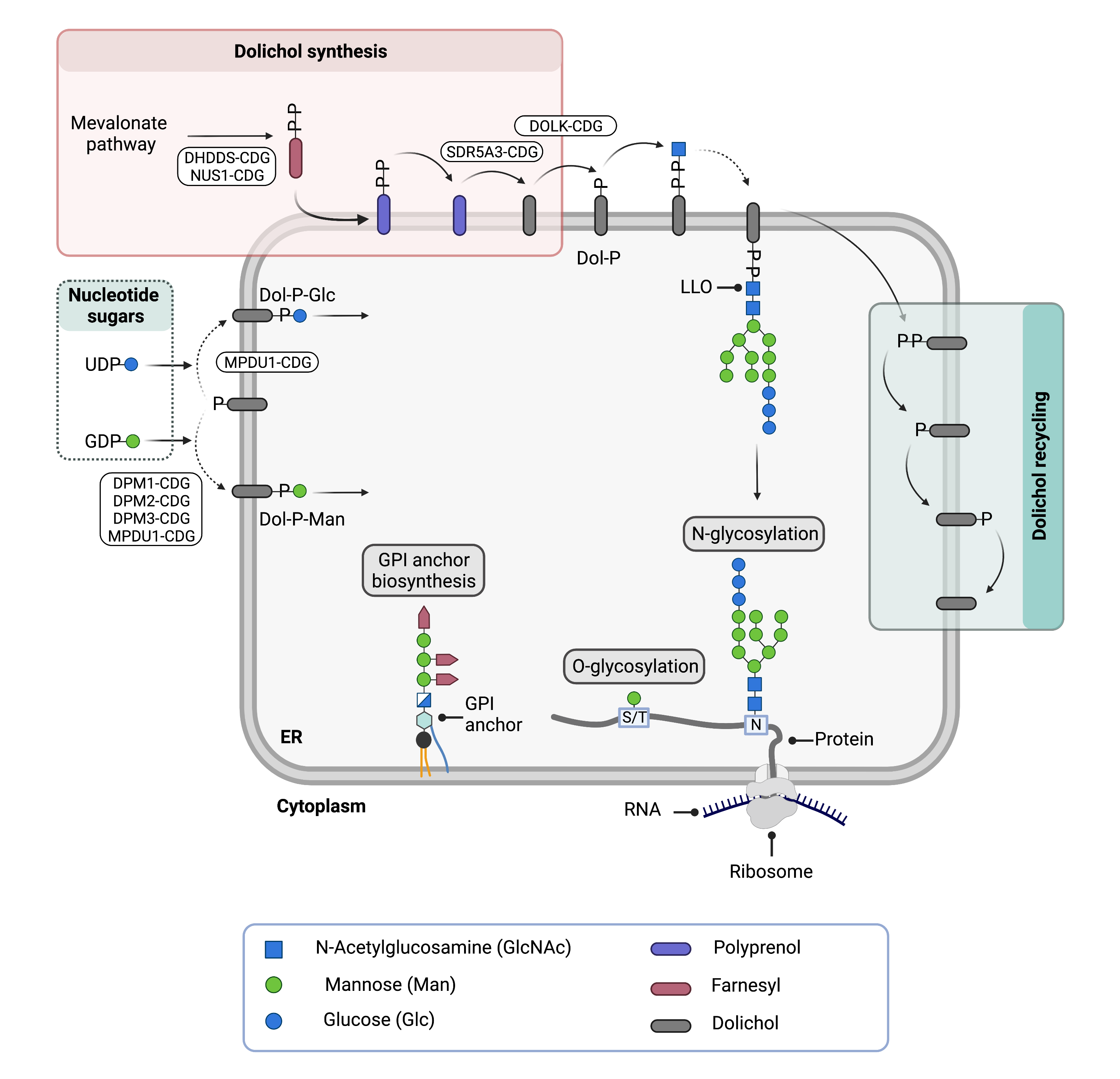
Figure 2. Overview of dolichol metabolism in different glycosylation pathways and associated CDG types.
Dolichol is generated through a series of enzymatic reactions and subsequently phosphorylated (Dol-P). Mannose or glucose are attached to Dol-P to generate activated sugars (Dol-P-Man; Dol-P-Glc) that function as sugar donors in several glycosylation pathways, including N- and O-glycosylation and GPI anchor biosynthesis. The lipid-linked-oligosaccharide (LLO) is built on Dol-P and the oligosaccharide is transferred to a protein in N-glycosylation.
Disorders of Nucleotide Sugar Synthesis and Transport
Disorders of nucleotide sugar synthesis and transport affect glycosylation pathways that use the corresponding nucleotide sugars. Nucleotide sugars are high-energy forms of monosaccharides which are necessary for enzymes to be able to transfer the monosaccharide to the protein, lipid or glycan. Nucleotide sugars are typically monosaccharides linked to phosphorylated nucleosides: UDP (uridine disphosphate), GDP (guanosine disphosphate) or CMP (cytidine monophosphate), generating monosaccharide-UDP, -GDP or -CMP, respectively. Nucleotide sugar synthesis is dependent on a series of enzyme reactions4. Generally, to generate nucleotide sugars, enzymes require a monosaccharide modified with a phosphate group (sugar phosphate) and a phosphorylated nucleoside, or nucleotide sugars can undergo interconversion (Figure 1)4,16.
Glycosylation occurs inside the ER and the Golgi, which are compartments that are surrounded by membranes that nucleotide sugars cannot pass through6. Nucleotide sugar transporters exist in the ER and Golgi membranes, and transport nucleotide sugars from the cytosol to the inside of these compartments. These nucleotide sugar transporters belong to a large group of proteins known as solute carrier (SLC) proteins, which move a wide array of substances across membranes (Figure 3)17. Consequently, defects in nucleotide sugar transporters can reduce availability of nucleotide sugars during glycosylation and can lead to CDG18.
Disorders of nucleotide sugar synthesis and transport include:
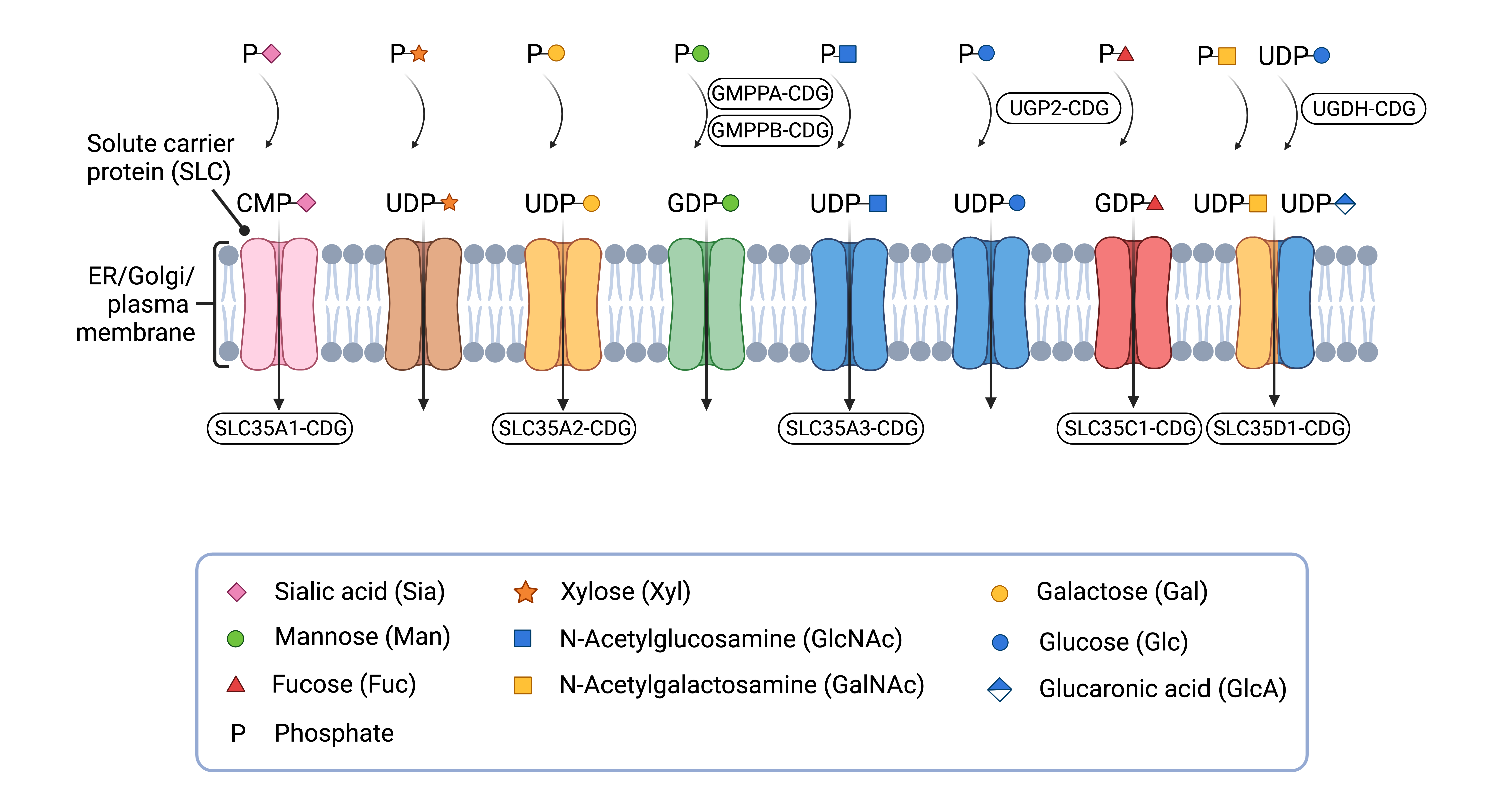
Figure 3. Overview of solute carrier (SLC) proteins and CDG caused by defects in nucleotide sugar synthesis or transport.
SLC proteins can transport nucleotide sugars into the different compartments of the cell, such as the ER and Golgi, for use in different glycosylation pathways.
Disorders of Vesicular Trafficking
CDG have been linked to a system that transports components between the ER and the Golgi, known as vesicular trafficking (Figure 4) 7. Cells use vesicles, enclosed structures surrounded by a membrane, to transport substances around the cell, including to different compartments like the ER and the Golgi. Vesicles are also used to move substances between the inside and the outside of a cell – this transportation system is known as vesicular trafficking. Vesicles are needed to shuttle different components of glycosylation between the ER and the Golgi, such as enzymes and proteins22,23. Furthermore, the Golgi is made up of different compartments, which allows different types of glycan modifications to occur in different sections of the Golgi, and vesicles are used to transport materials between these sub-compartments of the Golgi22.
Vesicles can be surrounded by coat protein complex (COP) proteins. The vesicles are able to attach to the Golgi membrane through the use of different proteins, such as the conserved oligomeric Golgi (COG) complex, SNAREs and tethering proteins, and CDG have been linked to defects in many of these proteins22,23.
Disorders of vesicle trafficking include:
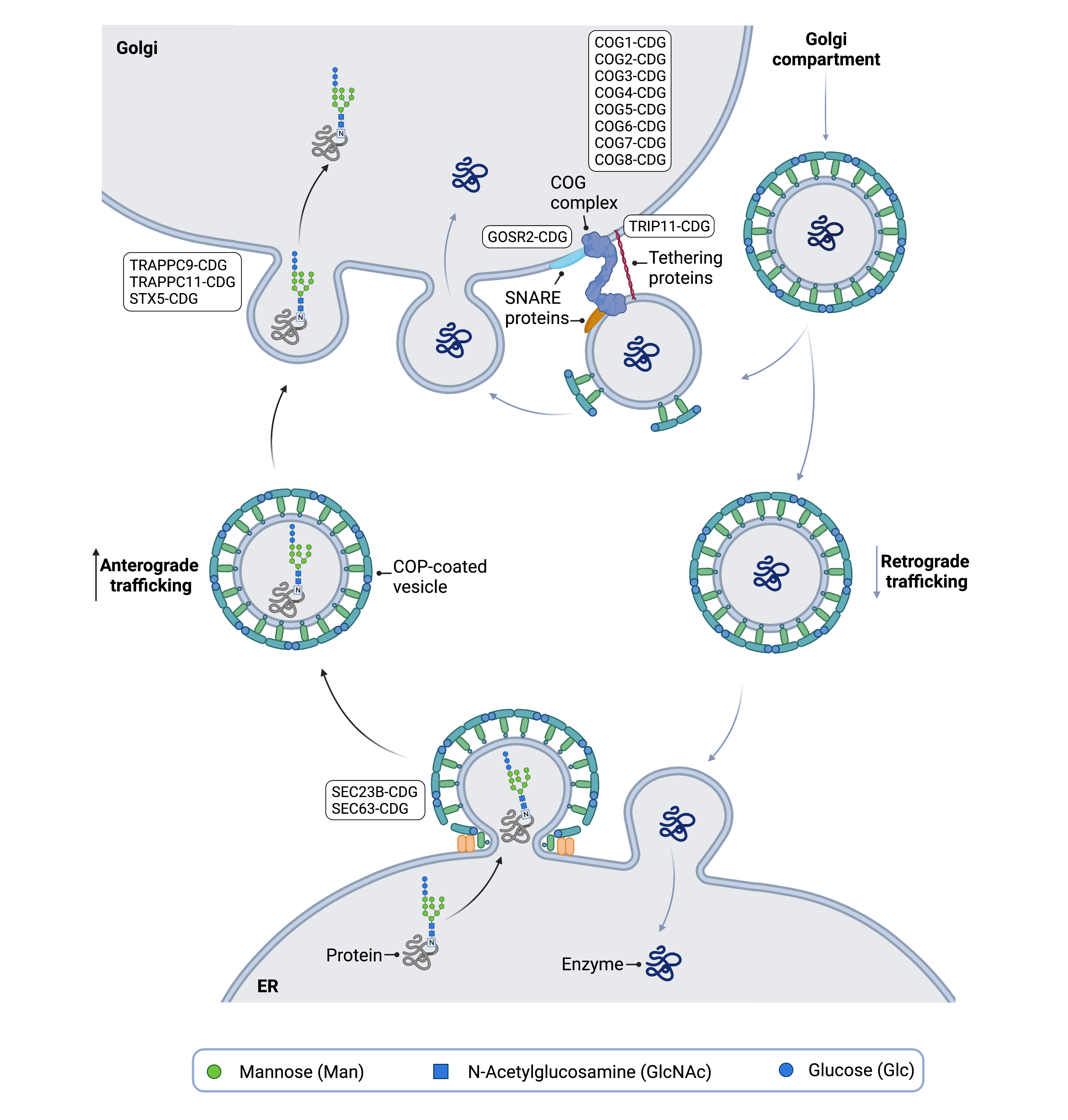
Figure 4. Overview of vesicular trafficking in glycosylation and associated CDG.
Vesicular trafficking uses vesicles to move substances used in glycosylation, such as proteins and enzymes, between different cell compartments. Disorders of vesicular trafficking may arise due to defects in proteins that form vesicle coat proteins and various proteins that tether and fuse vesicles to membranes.
Disorders of Golgi Homeostasis
The main functions of the Golgi are sorting glycolipids and proteins into vesicles for delivery to target destinations and glycosylation. Maintenance (homeostasis) of these functions is dependent on maintaining specific pH levels and ion concentrations in the Golgi compartments. The acidic pH of the Golgi is established and maintained by ion transport systems which include the vacuolar H+-ATPase (an enzyme complex that pumps protons across membranes) and transporters that move Cl-, Na+ and K+ ions across the membrane. Other ions such as Ca2+, Mg2+ and Mn2+ are important for glycosylation and cargo sorting in the Golgi and are maintained at high concentrations in Golgi lumen by a number of ion channels and transporters.
Defects in Golgi homeostasis, such as abnormal pH and ion levels are causative of some types of CDG8. Disorders in Golgi homeostasis can arise from defects in ion transporters and the vacuolar H+-ATPase complex (Figure 5)24. Abnormal pH and ion levels can reduce enzyme activity and lead to incorrect localization of glycosylation components22,25.
Disorders of Golgi homeostasis include:
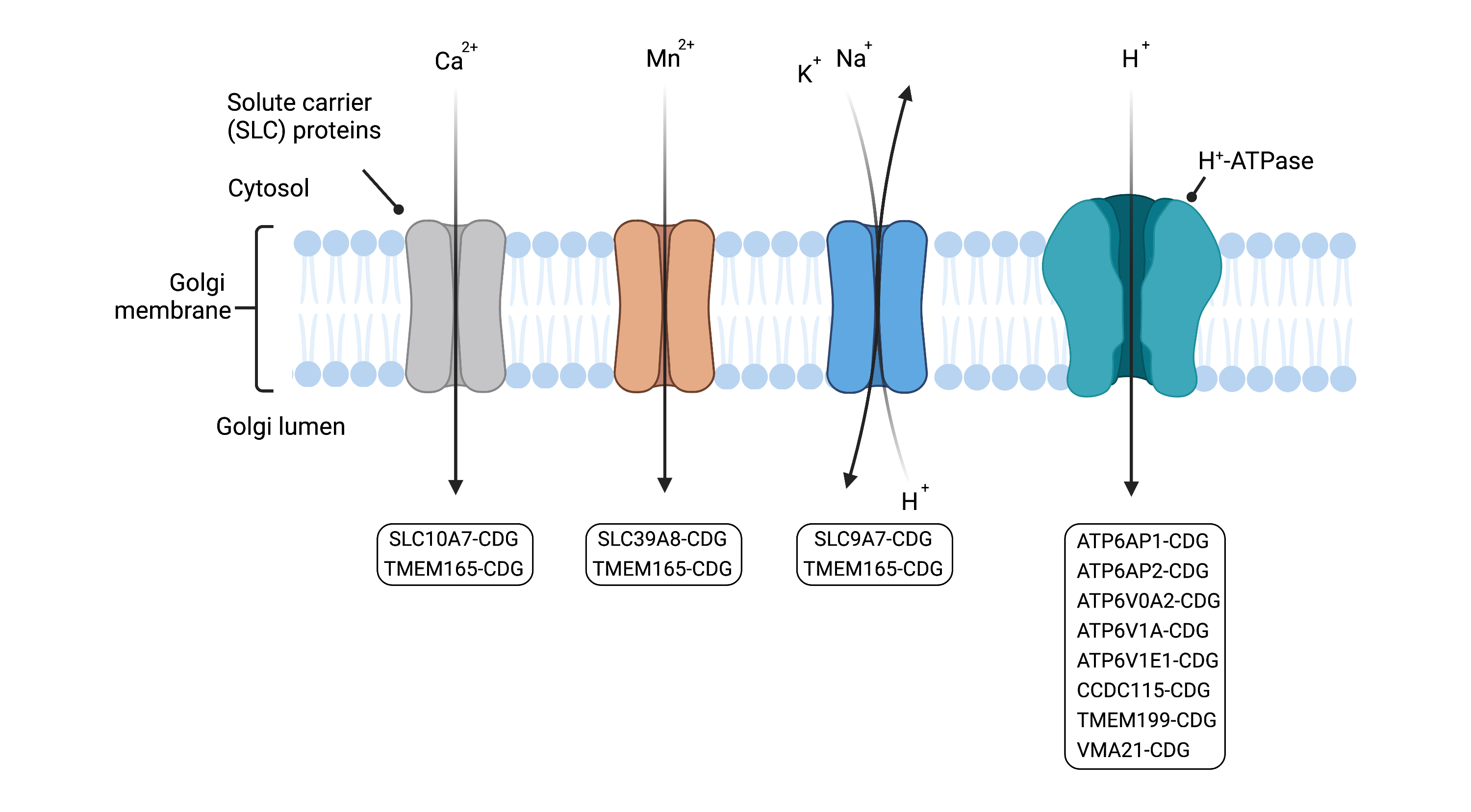
Figure 5. Overview of CDG caused by defects in Golgi homeostasis, arising from defects in ion transporters and vacuolar H+-ATPase.
Ion transporters move ions across the membrane of the Golgi and the vacuolar H+-ATPase maintains the pH of the Golgi. Defects in Golgi ion levels and pH can lead to reduced enzyme activity and glycosylation substances not being in the correct location.
Other Disorders of Multiple Glycosylation Pathways
Other disorders of multiple glycosylation pathways disrupt glycosylation through different processes than the ones described above. This includes CDG that arise due to defects in proteins (GET2-4) that facilitate targeting of membrane proteins to their correct location and a TGDS, an enzyme whose function is unknown but is linked to Catel-Manzke syndrome26.
Diagnosis
Disorders that affect more than one glycosylation pathway may result in changes that can be detected using the standard diagnostic tests and strategies employed for other CDG types. These may include analysis of transferrin (an abundant serum N-glycoprotein)27,28, apolipoprotein C-III (a protein with an O-glycan)29, total plasma or serum N- or O-glycans30,31, and alpha-dystroglycan (an abundant O-mannosylated glycoprotein)32,33. Importantly, in CDG that only affect one glycosylation pathway, such as N-glycosylation, one biochemical marker may be abnormal while others remain unchanged34,35. However, in diseases of multiple glycosylation pathways, it is possible to see abnormalities in multiple different tests (e.g. abnormal transferrin and apolipoprotein C-III)36. If symptoms are highly suggestive of a problem caused by a defect in a particular gene, the activity of the corresponding enzyme can also be measured for some CDG37. Ultimately, these markers vary between disease and individual and genetic testing is required for definitive diagnosis38.
Transferrin Analysis
Analysis of serum transferrin, also referred to as transferrin isoform analysis or carbohydrate deficient transferrin (CDT) analysis, may be used to help identify defects in N-glycosylation. Serum transferrin normally has two N-glycans with a total of four sialic acid sugars referred to as tetrasialotransferrin39. In CDG affecting N-glycosylation, transferrin usually has altered or reduced glycosylation, with changes to the number of sialic acids in the N-glycans or chains missing entirely35,40. There are several methods to analyse transferrin, but differences in the abundance of transferrin isoforms is often measured using transferrin isoelectric focusing (TIEF) or mass spectrometry41. N-linked CDG typically result in glycosylation patterns on transferrin that can be used to determine whether the CDG impacts the early steps (Type I pattern) or later steps (Type II pattern) of N-glycoprotein synthesis35. In CDG that affect multiple pathways, the analysis of transferrin isoforms may also reveal a pattern that is neither a Type I nor Type II, known as a mixed type I/II pattern38.
Apolipoprotein C-III Analysis
Apolipoprotein C-III (apo C-III) is a glycoprotein found in the blood that has a single O-linked glycan and is used to identify disorders of O-linked glycosylation42. Apolipoprotein C-III exists in three isoforms which differ in their number of sialic acid residues, including zero (apo C-III0), one (apo C-III1) or two (apo C-III2) sialic acid residues43. CDG that arise from defects in O-glycosylation and defects in Golgi homeostasis have increased levels of apo C-III0 and/or apo C-III144. These can be detected by different methods, such as isoelectric focusing, two-dimensional gel electrophoresis, capillary electrophoresis and mass spectrometry45,46.
Alpha-dystroglycan Analysis
Alpha-dystroglycan is an O-mannosylated subunit of the dystroglycan protein that is expressed in muscle and brain tissue32,33. Inadequate or incorrect glycosylation of alpha-dystroglycan is typically seen in CDG involving O-mannosylation but can also be caused by CDG affecting dolichol metabolism47. Alpha-dystroglycan can be analyzed with specialized staining techniques that use antibodies that bind to glycosylated alpha-dystroglycan48.
Total Glycan Analysis
Chemical analysis of the total N- and O-glycans present in patient plasma or serum may be used as part of diagnosis of CDG which affect multiple pathways49.
Genetic Testing
Genetic testing is often required to definitively diagnose CDG involving multiple pathways, and may include targeted sequencing of specific genes, use of CDG gene panels that test for known CDG, whole genome sequencing (WGS), and the sequencing of all expressed genes via whole exome sequencing (WES)50–53
References
- Schjoldager, K. T., Narimatsu, Y., Joshi, H. J. & Clausen, H. Global view of human protein glycosylation pathways and functions. Nature Reviews Molecular Cell Biology 21, (2020).
- Reily, C., Stewart, T. J., Renfrow, M. B. & Novak, J. Glycosylation in health and disease. Nature Reviews Nephrology 15, (2019).
- Taylor, C. M. Glycopeptides and glycoproteins: Focus on the glycosidic linkage. Tetrahedron vol. 54 (1998).
- Mikkola, S. Nucleotide Sugars in Chemistry and Biology. Molecules 25, (2020).
- Rini, J. M. & Esko, J. D. Glycosyltransferases and Glycan-Processing Enzymes. Essentials of Glycobiology (2015).
- Hadley, B. et al. Structure and function of nucleotide sugar transporters: Current progress. Computational and Structural Biotechnology Journal 10, (2014).
- Matalonga, L. et al. Mutations in TRAPPC11 are associated with a congenital disorder of glycosylation. Human Mutation 38, (2017).
- Ondruskova, N., Cechova, A., Hansikova, H., Honzik, T. & Jaeken, J. Congenital disorders of glycosylation: Still “hot” in 2020. Biochimica et Biophysica Acta (BBA) - General Subjects 1865, (2021).
- Ng, B. G. et al. Pathogenic Variants in Fucokinase Cause a Congenital Disorder of Glycosylation. The American Journal of Human Genetics 103, (2018).
- van Karnebeek, C. D. M. et al. NANS-mediated synthesis of sialic acid is required for brain and skeletal development. Nature Genetics 48, (2016).
- Eklund, E. A., Bode, L. & Freeze, H. H. Diseases Associated with Carbohydrates/Glycoconjugates*. in Comprehensive Glycoscience (Elsevier, 2007). doi:10.1016/B978-044451967-2/00098-2.
- Freeze, H. H. & Elbein, A. D. Chapter 4 Glycosylation Precursors. Essentials of Glycobiology 2nd Edition (2009).
- Denecke, J. & Kranz, C. Hypoglycosylation due to dolichol metabolism defects. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1792, (2009).
- Cantagrel, V. & Lefeber, D. J. From glycosylation disorders to dolichol biosynthesis defects: a new class of metabolic diseases. Journal of Inherited Metabolic Disease 34, (2011).
- Buczkowska, A., Swiezewska, E. & Lefeber, D. J. Genetic defects in dolichol metabolism. Journal of Inherited Metabolic Disease 38, (2015).
- Freeze, H. H., Schachter, H. & Kinoshita, T. Genetic disorders of glycosylation. in Essentials of Glycobiology (eds. Varki, A., Cummings, R. & Esko, J.) ( Cold Spring Harbor Laboratory Press, 2017).
- Pizzagalli, M. D., Bensimon, A. & Superti‐Furga, G. A guide to plasma membrane solute carrier proteins. The FEBS Journal 288, (2021).
- Ng, B. G. et al. Encephalopathy caused by novel mutations in the CMP-sialic acid transporter, SLC35A1. American Journal of Medical Genetics Part A 173, (2017).
- Chou, J. Y. & Mansfield, B. C. The SLC37 Family of Sugar-Phosphate/Phosphate Exchangers. in (2014). doi:10.1016/B978-0-12-800223-0.00010-4.
- Chou, J. Y., Sik Jun, H. & Mansfield, B. C. The SLC37 family of phosphate-linked sugar phosphate antiporters. Molecular Aspects of Medicine 34, (2013).
- Wilson, M. P. et al. SLC37A4‐CDG: Second patient. JIMD Reports 58, (2021).
- Linders, P. T. A., Peters, E., ter Beest, M., Lefeber, D. J. & van den Bogaart, G. Sugary Logistics Gone Wrong: Membrane Trafficking and Congenital Disorders of Glycosylation. International Journal of Molecular Sciences 21, (2020).
- Witkos, T. M. et al. GORAB scaffolds COPI at the trans-Golgi for efficient enzyme recycling and correct protein glycosylation. Nature Communications 10, (2019).
- Pamarthy, S., Kulshrestha, A., Katara, G. K. & Beaman, K. D. The curious case of vacuolar ATPase: regulation of signaling pathways. Molecular Cancer 17, (2018).
- Raynor, A. et al. Normal transferrin patterns in congenital disorders of glycosylation with Golgi homeostasis disruption: apolipoprotein C-III at the rescue! Clinica Chimica Acta 519, (2021).
- Tambe, M. A. et al. Mutations in GET4 disrupt the transmembrane domain recognition complex pathway. Journal of Inherited Metabolic Disease 43, (2020).
- Mohamed, M. et al. Clinical and diagnostic approach in unsolved CDG patients with a type 2 transferrin pattern. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1812, (2011).
- Gkouvatsos, K., Papanikolaou, G. & Pantopoulos, K. Regulation of iron transport and the role of transferrin. Biochimica et Biophysica Acta - General Subjects vol. 1820 188–202 (2012).
- Wada, Y. & Okamoto, N. Apolipoprotein C-III O-glycoform profiling of 500 serum samples by matrix-assisted laser desorption/ionization mass spectrometry for diagnosis of congenital disorders of glycosylation. Journal of Mass Spectrometry 56, (2021).
- Abu Bakar, N. et al. Intact transferrin and total plasma glycoprofiling for diagnosis and therapy monitoring in phosphoglucomutase-I deficiency. Translational Research 199, (2018).
- Park, J. H. et al. N-glycome analysis detects dysglycosylation missed by conventional methods in SLC39A8 deficiency. Journal of Inherited Metabolic Disease 43, (2020).
- van Tol, W. et al. A mutation in mannose-phosphate-dolichol utilization defect 1 reveals clinical symptoms of congenital disorders of glycosylation type I and dystroglycanopathy. JIMD Reports 50, (2019).
- Nickolls, A. R. & Bönnemann, C. G. The roles of dystroglycan in the nervous system: Insights from animal models of muscular dystrophy. DMM Disease Models and Mechanisms vol. 11 (2018).
- Chang, I. J., He, M. & Lam, C. T. Congenital disorders of glycosylation. Annals of Translational Medicine 6, (2018).
- Lefeber, D. J., Morava, E. & Jaeken, J. How to find and diagnose a CDG due to defective N-glycosylation. Journal of Inherited Metabolic Disease 34, (2011).
- Bogdańska, A. et al. Clinical, biochemical and molecular phenotype of congenital disorders of glycosylation: long-term follow-up. Orphanet Journal of Rare Diseases 16, (2021).
- Grünert, S. C. et al. Unsuccessful intravenous D-mannose treatment in PMM2-CDG. Orphanet Journal of Rare Diseases 14, (2019).
- Lipiński, P. & Tylki-Szymańska, A. Congenital Disorders of Glycosylation: What Clinicians Need to Know? Frontiers in Pediatrics vol. 9 (2021).
- Wopereis, S. et al. Patients with unsolved congenital disorders of glycosylation type II can be subdivided in six distinct biochemical groups. Glycobiology 15, 1312–1319 (2005).
- Denecke, J. et al. Congenital disorder of glycosylation type Id: Clinical phenotype, molecular analysis, prenatal diagnosis, and glycosylation of fetal proteins. Pediatric Research 58, (2005).
- van Scherpenzeel, M., Willems, E. & Lefeber, D. J. Clinical diagnostics and therapy monitoring in the congenital disorders of glycosylation. Glycoconjugate Journal 33, (2016).
- Taskinen, M. R. & Borén, J. Why Is Apolipoprotein CIII Emerging as a Novel Therapeutic Target to Reduce the Burden of Cardiovascular Disease? Current Atherosclerosis Reports vol. 18 (2016).
- Yassine, H. N. et al. The association of human apolipoprotein C-III sialylation proteoforms with plasma triglycerides. PLoS ONE 10, (2015).
- Raynor, A. et al. Normal transferrin patterns in congenital disorders of glycosylation with Golgi homeostasis disruption: apolipoprotein C-III at the rescue! Clinica Chimica Acta 519, (2021).
- Ruel, C. et al. A capillary zone electrophoresis method for detection of Apolipoprotein C-III glycoforms and other related artifactually modified species. Journal of Chromatography A 1532, (2018).
- Yen-Nicolaÿ, S. et al. MALDI-TOF MS applied to apoC-III glycoforms of patients with congenital disorders affecting O-glycosylation: Comparison with two-dimensional electrophoresis. Proteomics - Clinical Applications 9, (2015).
- Lefeber, D. J. et al. Deficiency of Dol-P-Man Synthase Subunit DPM3 Bridges the Congenital Disorders of Glycosylation with the Dystroglycanopathies. American Journal of Human Genetics 85, (2009).
- Martinez, H. R. et al. Novel cardiovascular findings in association with a POMT2 mutation: Three siblings with α-dystroglycanopathy. European Journal of Human Genetics 22, (2014).
- Xia, B. et al. Serum N-glycan and O-glycan analysis by mass spectrometry for diagnosis of congenital disorders of glycosylation. Analytical Biochemistry 442, (2013).
- Francisco, R. et al. The challenge of CDG diagnosis. Molecular Genetics and Metabolism 126, 1–5 (2019).
- Timal, S. et al. Gene identification in the congenital disorders of glycosylation type i by whole-exome sequencing. Human Molecular Genetics 21, 4151–4161 (2012).
- Bruneel, A., Cholet, S., Tran, N. T., Mai, T. D. & Fenaille, F. CDG biochemical screening: Where do we stand? Biochimica et Biophysica Acta - General Subjects 1864, (2020).
- Chang, I. J., He, M. & Lam, C. T. Congenital disorders of glycosylation. Annals of Translational Medicine 6, 1–13 (2018).
- Khosrowabadi, E., and Kellokumpu, S. Golgi pH and Ion Homeostasis in Health and Disease. In: Reviews of Physiology, Biochemistry and Pharmacology (2020).