
Lay Summary
ALG2-CDG, formerly known as CDG-Ii, is a rare inherited condition that affects many systems in the body. To date, 9 cases have been reported in the medical literature. ALG2-CDG is classified as a disorder of N-linked protein glycosylation. ALG2-CDG is caused when an individual has mutations in both copies of the ALG2 gene, which provides instructions for making an enzyme that attaches the simple sugar mannose to growing sugar chains during N-glycosylation. ALG2-CDG patients may have a severe presentation, characterized by neurological abnormalities including developmental delay, cognitive impairment, low muscle tone, epilepsy, ophthalmological problems and dysmorphic features, or a mild presentation primarily consisting of a neuromuscular transmission disorder associated with muscle weakness and fatigue; the mild presentation is also known as ALG2 congenital myasthenic syndrome (ALG2-CMS). Screening tests are available for ALG2-CDG, but a definitive diagnosis is achieved through genetic testing. Treatment is primarily is focused on the management of specific symptoms and preventing complications. Acetylcholinesterase inhibitors have been used to treat muscle-related symptoms in some patients.
Overview
Alpha-1,3-mannosyltransferase 2 congenital disorder of glycosylation (ALG2-CDG) is a rare autosomal recessive genetic disorder. The ALG2 gene encodes an enzyme that is responsible for sequentially attaching the second and third mannose residues to the growing oligosaccharide on the lipid carrier dolichol; this is the third and fourth step in the synthesis of the lipid-linked oligosaccharide (LLO) in the endoplasmic reticulum (ER)2 (Figure 1). LLO synthesis is a precursor step to N-glycosylation, which is the process by which sugar chains (glycans) are added to the amino acid asparagine in some proteins. Mutations to ALG2 result in an enzyme with reduced catalytic activity, where mannose transfer is reduced or absent. Deficiency in the ALG2 enzyme results in the incomplete assembly of the LLO, leading to insufficient N-glycosylation of glycoproteins2,4.
The first reported case of ALG2-CDG was in 20031 and there are 91–3 confirmed cases to date. Symptoms of ALG2-CDG typically begin in infancy, although they may not be present at birth. Characteristic presentations of ALG2-CDG include neurological symptoms, ophthalmological problems, and dysmorphic features1,2.
Several genes associated with CDG (ALG2, ALG14, DPAGT1, GFPT1) are also associated with congenital myasthenic syndrome (CMS), a group of neuromuscular transmission disorders. The clinical presentation of ALG2-associated CMS (ALG2-CMS) is less severe than ALG2-CDG and is characterized by muscle weakness and fatigue beginning in childhood5,6. A diagnosis of ALG2-CDG can be determined through transferrin analysis, but a definitive diagnosis can only be determined through genetic testing. There are currently no approved treatments for ALG2-CDG, however some patients diagnosed with CMS symptoms have seen improvements in muscle related symptoms with acetylcholinesterase inhibitors5,6.
Synonyms
- CDG-1I
- CDG-Ii
- CDG syndrome type Ii
- Congenital disorder of glycosylation type Ii
- Carbohydrate deficient glycoprotein syndrome type Ii
- Alpha-1,3-mannosyltransferase 2 congenital disorder of glycosylation
- Mannosyltransferase 2 deficiency
- Myasthenic syndrome, congenital, 14, with tubular aggregates
Inheritance
ALG2-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The ALG2 gene encodes the enzyme alpha-1,3-mannosyltransferase (ALG2). ALG2 is located in the ER membrane where it has a role in the assembly of the LLO, the oligosaccharide precursor used in protein N-glycosylation7. The ALG2 enzyme catalyzes the fourth and fifth steps in LLO synthesis: the sequential addition of two mannose residues to the growing oligosaccharide chain attached to the lipid carrier, dolichol pyrophosphate (Dol-PP)1,2,4,5.
LLO synthesis
N-glycosylation is the process by which an oligosaccharide is attached to the nitrogen atom of asparagine residues on proteins. N-glycosylation is initiated in the ER and begins with the synthesis of the LLO8. The LLO is comprised of a 14-sugar oligosaccharide, which is sometimes referred to as the N-glycan precursor oligosaccharide, attached to the lipid carrier dolichol pyrophosphate (Dol-PP). The 14-sugar glycan chain is made up of 2 N-acetylglucosamine, 9 mannose, and 3 glucose residues. Once assembled, the oligosaccharide is transferred en bloc to proteins and undergoes further processing in the ER and Golgi9. Once the oligosaccharide is attached to a protein, it is referred to as an N-glycan.
LLO synthesis is carried out by a series of enzymes encoded by the DPAGT1 and ALG genes and can be divided into two phases: Phase I and Phase II 9(Figure 1).
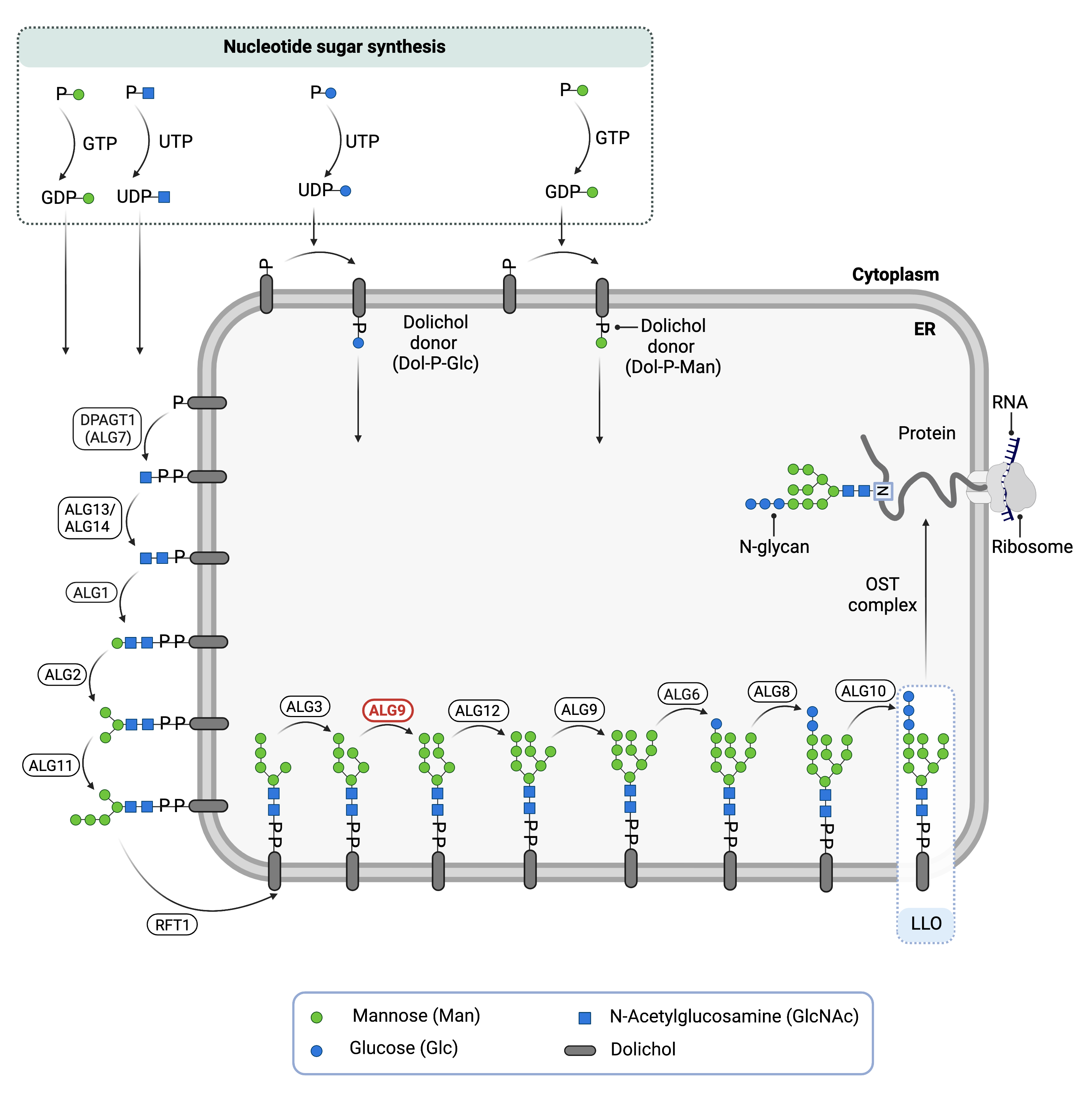
Figure 1.Role of ALG2 in glycosylation.
ALG2 is an enzyme (alpha-1,3-mannosyltransferase) involved in synthesizing the lipid-linked oligosaccharide (LLO) in N-glycosylation. ALG2 adds two mannose residues to the LLO on the cytosolic side of the endoplasmic reticulum membrane.
Phase I
Phase I of LLO synthesis takes place on the cytoplasmic side of the ER. It begins with the sequential attachment of two N-Acetylglucosamine (GlcNAc2) and five mannose (Man5) residues to Dol-PP by several ER enzymes. The ALG2 enzyme catalyzes the transfer of two mannose residues from the nucleotide sugar GDP-mannose onto the growing LLO, generating Dol-PP-GlcNAc2Man3.The subsequent and mannose sugars are transferred from GDP-mannose onto the growing LLO. The intermediate structure, Dol-PP-GlcNAc2Man5, is translocated across the ER membrane into the lumen by the RFT1 enyzme8,9.
Phase II
Phase II of LLO synthesis takes place in the ER lumen. Once in the lumen, four mannose residues followed by three glucose (Glc3) residues are added to the intermediate structure, generating the complete LLO, Dol-PP-GlcNAc2Man9Glc3. Mannose and glucose are transferred from glycosyl donors Dol-P-mannose and Dol-P-glucose, which are formed on the cytoplasmic side of the ER and must also be flipped across the ER membrane8.
Once assembled, the oligosaccharide is transferred en bloc from Dol-PP to asparagine residues of newly synthesized proteins via the enzyme complex oligosaccharyltransferase (OST), resulting in N-glycosylation of the protein13. The activity of OST is highly specific for the completely assembled 14-sugar oligosaccharide, GlcNAc2Man9Glc3.
Disease Mechanism
Mutations in the ALG2 gene lead to the production of an abnormal enzyme with reduced or no activity, thereby disrupting the transfer of mannose residues onto the growing LLO. As a result, LLO synthesis is incomplete and the transfer of its glycan component to proteins is impaired, resulting in N-glycoproteins that are insufficiently glycosylated (hypoglycosylation) and accumulation of Dol-PP-GlcNAc2Man1 and Dol-PP-GlcNAc2Man25. Since N-glycans are important for the proper function of many processes in the body, ALG2-CDG affects multiple systems.
In ALG2-CMS, mutations to ALG2 severely reduces ALG2 enzyme expression in muscles; it is essential along with DPAGT1 and ALG14 for the proper functioning of the neuromuscular junction5.
Mutations
The ALG2 gene is located on Chromosome 9 (9p31.1) and only 4 mutations have been reported thus far11. The most severe mutation reported is a heterozygous mutation with a deletion (Δ1040G) and single nucleotide substitution (G393T)1.
Signs & Symptoms
Clinical Presentation
Individuals with ALG2-CDG typically have a multisystemic presentation or a mild presentation that primarily involves muscle weakness (ALG2-CMS).
ALG2-CDG
Individuals with ALG2-CDG with typically develop signs and symptoms during infancy. ALG2-CDG is primarily characterized by a multi-system disorder with mild to severe neurological disorder. The characteristic clinical presentations of ALG2-CDG include1,3
- Neurological – global developmental delay, intellectual disability, muscle weakness and low muscle tone (hypotonia), proximal muscle weakness, lack of coordination and muscle control (ataxia), epilepsy, increased reflexes (hyperreflexia), and disorganized brain electrical activity (hypsarrhythmia)
- Ophthalmological – impaired vision, cataracts, involuntary rapid eye movement (nystagmus)
- Dysmorphic features – notched pupil (coloboma), enlarged liver, small head (microcephaly)
ALG2-CMS
Individuals with ALG2-CMS typically present with symptoms in childhood which is primarily characterized by fatigable muscle weakness. The characteristic clinical presentations of ALG2-CMS include5,6,11:
- Neurological symptoms – severely affected proximal and distal limb muscle weakness, limb girdle pattern of muscle weakness, mildly affected facial, ocular, bulbar, and respiratory muscles
Patients with CMS do not display the other multi-system symptoms associated with ALG2-CDG.
Biochemical Abnormalities
ALG2-CDG
Biochemical abnormalities observed in individuals with ALG2-CDG include blood clotting (coagulation) abnormalities, and elevated liver enzymes (serum transaminases)2.
ALG2-CMS
Patients with ALG2-CMS have also shown decreased or absent serum acetylcholine receptor and muscle-specific tyrosine kinase antibodies6.
Classification
ALG2-CDG is classified as a disorder of N-linked protein glycosylation.
Under the former CDG classification system, ALG2-CDG is classified as a Type 1 CDG which arise due to defects in the production of lipid-linked oligosaccharides or their transfer to proteins.
Diagnosis
Although diagnosis of ALG2-CDG may be suspected based on presentation of symptoms and a detailed patient history, direct molecular genetic testing is the only definitive diagnostic test. Screening in suspected patients begins with a blood test to analyze serum transferrin. Profiling of total serum N-glycans by mass spectrometry and LLO analysis in patient fibroblasts may also be carried out. Specialized testing on muscle and nerves is typically carried out when ALG2-CMS is suspected.
Transferrin Analysis
Individuals with ALG2-CDG sample show a type 1 pattern by mass spectrometry analysis of transferrin 2. Type 1 patterns are observed in CDG that arise due to defects in LLO assembly and are characterized by a decrease in tetrasialo-transferrin and an increase in di-sialo and a-sialo transferrin isoforms.
Total N-glycan Analysis
Total N-glycan profiling of ALG2-CDG patient serum reveals an increase in hyposialylated and fucosylated glycoforms as well as the presence of high-mannose, hybrid, and truncated glycoforms2.
LLO Analysis
LLO analysis of ALG2-CDG patient skin fibroblasts have an accumulation of Dol-PP-GlcNAc2Man1 and Dol-PP-GlcNAc2Man21.
Muscle Biopsy
Muscle biopsy of patients with ALG2-CMS show myopathic features, ragged red fiber, and accumulation of mitochondria on the sub-sarcolemma11.
Single Fiber Electromyography (EMG)
For patients with ALG2-CMS, single fiber EMG or nerve stimulation tests can be used to investigate the activity of individual muscle fibers and detect abnormal signalling between nerves and muscles6.
Prognosis
Prognosis of ALG2-CDG may vary depending on the severity of an individual’s symptoms. Most patients reported live into adulthood with slow progression and muscle deterioration, becoming wheelchair dependent5.
Management
Management of symptoms may include combinations of physical therapy, occupational therapy, speech or vision therapy and palliative measures.
Therapies
It has been suggested that acetylcholinesterase inhibitors may improve muscle-related symptoms in patients with myasthenia5. Treatment for other symptoms is focused on management of symptoms and prevention of complications.
Research Models
Several ALG2 research models have been generated including yeast, fly, worm, mouse model organisms and patient-derived fibroblasts.
Yeast (S. cerevisiae)
Alg2 was first characterized in yeast and the role of the alg2 enzyme in N-glycosylation. Alg2 mutants (alg2-1) had temperature sensitive growth and accumulation of Man2GlcNAc2-PP-Dol12. The mutant alg2-1 sequence was used to identify the human ortholog when investigating the first reported ALG2-CDG patient, as it was known to accumulate similar Dol-PP linked oligosaccharides observed in patient skin fibroblasts1.
Fly (D. melanogaster)
Drosophila gene Alg2 (Alg2, CG1291, FBgn0035401) is orthologous to human ALG2, also encoding the alpha-1,3-mannosyltransferase enzyme. It is a potential model for the human disease ALG2-CDG (FlyBase).
Fish (O. latipes)
Oryzias latipes were used to model developmental consequences of ALG2 mutations seen in ALG2-CDG. Hypomorphic mutations affected protein abundance and phenotypically resulted in multisystemic problems including in neuronal tissue and facial skeleton abnormalities4.
Mouse (M. musculus)
Homozygous Alg2-/- knockout
Homozygous Alg2 knockout mice are embryonic lethal before organs develop (E9.5), and display preweaning lethality with complete penetrance (early adult) (IMPC, MGI).
Heterozygous Alg2+/- knockout
Heterozygous mutations affect homeostasis and metabolism, phenotypically presenting with decreased fasting circulating glucose levels and increased circulating insulin levels, surviving to early adulthood IMPC, MGI).
Immunofluorescence labelling of mouse neuromuscular junction
Localization of Alg2 in mouse muscle support hypotheses for the importance of ALG2 and other N-linked glycosylation proteins at the neuromuscular junction. Alg2 and Alg14 are both localized at the neuromuscular junction and endplate regions in mouse muscle, consistent with -CMS and -CDG symptoms associated with muscle weakness and affected neuromuscular transmission5.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including ALG2-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Publications
ALG2-CDG Scientific Articles on PubMed
Additional Resources
References
- Thiel, C. et al. A New Type of Congenital Disorders of Glycosylation (CDG-Ii) Provides New Insights into the Early Steps of Dolichol-linked Oligosaccharide Biosynthesis. Journal of Biological Chemistry 278, 22498–22505 (2003).
- Papazoglu, G. M. et al. Mass spectrometry glycophenotype characterization of ALG2-CDG in Argentinean patients with a new genetic variant in homozygosis. Glycoconjugate Journal 38, 191–200 (2021).
- Asteggiano, C. G. et al. Ten years of screening for congenital disorders of glycosylation in Argentina: case studies and pitfalls. Pediatric Research 84, 837–841 (2018).
- Gücüm, S. et al. A patient-based medaka alg2 mutant as a model for hypo- N -glycosylation. Development 148, (2021).
- Cossins, J. et al. Congenital myasthenic syndromes due to mutations in ALG2 and ALG14. Brain 136, 944–956 (2013).
- Ehrstedt, C., Liu, W.-W., Frykholm, C., Beeson, D. & Punga, A. R. Novel pathogenic ALG2 mutation causing congenital myasthenic syndrome: a case report. Neuromuscular Disorders (2021)
- Dong, Y. Y. et al. Structures of DPAGT1 Explain Glycosylation Disease Mechanisms and Advance TB Antibiotic Design. Cell 175, 1045-1058.e16 (2018).
- Bieberich, E. Synthesis, Processing, and Function of N-glycans in N-glycoproteins. in (2014).
- Stanley, P., Taniguchi, N. & Aebi, M. N-glycans. in Essentials of Glycobiology [Internet] (eds. Varki, A. et al.) (Cold Spring Harbor Laboratory Press, 2017).
- Harada, Y., Ohkawa, Y., Kizuka, Y. & Taniguchi, N. Oligosaccharyltransferase: A gatekeeper of health and tumor progression. International Journal of Molecular Sciences vol. 20 (2019).
- Monies, D. M. et al. Clinical and pathological heterogeneity of a congenital disorder of glycosylation manifesting as a myasthenic/myopathic syndrome. Neuromuscular Disorders 24, 353–359 (2014).
- Jackson, B. J., Kukuruzinska, M. A. & Robbins, P. Biosynthesis of asparagine-linked oligosaccharides in Saccharomyces cerevisiae: the alg2 mutation. Glycobiology 3, 357–364 (1993).