
Lay Summary
ALG14-CDG is a rare inherited condition that affects many parts of the body. To date, 12 patients have been reported in the medical literature. ALG14-CDG is classified as a disorder of N-linked protein glycosylation. ALG14-CDG is caused when an individual as mutations in both copies of their ALG14 gene, which provides instructions for making an enzyme that attaches the sugar N-acetylglucosamine to growing sugar chains during N-glycosylation. Mutations in the ALG14 gene cause proteins to be under glycosylated. ALG14-CDG patients may have a severe presentation, characterized my neurological abnormalities, intellectual disability, epilepsy, and dysmorphic features, or a mild presentation primarily consisting of a neuromuscular transmission disorder associated with muscle weakness and fatigue; the mild presentation is also known as ALG14 congenital myasthenic syndrome (ALG14-CMS). Screening tests typically used for CDG are not reliable in ALG14-CDG patients; diagnosis is achieved through genetic testing. Mild cases of ALG14-CDG have seen some long-term success with cholinesterase inhibitors as treatment and other patients have seen temporary improvement when being treated with Pyridostigmine.
Overview
ALG14-CDG is a rare autosomal recessive genetic disorder. The ALG14 (asparagine-linked glycosylation 14 homolog) gene encodes a subunit of the UDP-N-acetylglucosamine transferase enzyme responsible for adding the second N-acetylglucosamine residue during synthesis of the lipid linked oligosaccharide (LLO) in the endoplasmic reticulum (ER)1. LLO is a precursor step to N-glycosylation. Deficiency in the ALG14 enzyme results in incomplete assembly of the LLO, leading to insufficient N-glycosylation of glycoproteins1.
The first reported case of ALG14-CDG was in 20122 and to date, 12 cases have been reported in the literature 1–4. Symptoms being at infancy and the characteristic presentations of ALG14-CDG is congenital myasthenic syndrome, joint contractures of the knees, elbows, and ankles, and dysmorphic features1.
Several genes associated with CDG (DPAGT1, ALG2, ALG14, GFPT1) are also associated with congenital myasthenic syndrome (CMS), a group of neuromuscular transmission disorders. The clinical presentation of ALG14-associated CMS (ALG14-CMS) is less severe than ALG14-CDG and is characterized by muscle weakness and fatigue beginning in childhood. A diagnosis can only be determined through molecular genetic testing, as screening tests typically used for CDG-I patients are normal for the majority of ALG14-CDG patients. ALG14-CMS and mild cases of ALG14-CDG have seen some long-term success with cholinesterase inhibitors as treatment and some patients have seen temporary improvement when being treated with Pyridostigmine1.
Synonyms
- Congenital myasthenic syndrome without tubular aggregates-15; CMSWTA
- Myasthenic syndrome, congenital, 15; CMS15
Inheritance
ALG14-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The ALG14 gene encodes the ER membrane-bound subunit of the UDP-N-acetylglucosamine (GlcNAc) transferase enzyme (ALG14) 1,5. The other subunit of UDP-GlcNAc transferase is encoded by the ALG13 gene. UDP-GlcNAc transferase is a glycosyltransferase enzyme that is involved in the in the assembly of the LLO, a precursor for protein N-glycosylation1,5. UDP-GlcNAc glycosyltransferase catalyzes the transfer of the second GlcNAc residue from nucleotide sugar UDP-GlcNAc to the growing oligosaccharide precursor prior to its attachment to a protein.
LLO synthesis
N-glycosylation is the process by which an oligosaccharide is attached the nitrogen atom of asparagine residues on proteins. N-glycosylation is initiated in the ER and begins with the synthesis of the LLO 6. The LLO is comprised of a 14-sugar oligosaccharide, which is sometimes referred to as the N-glycan precursor, attached to the lipid carrier dolichol pyrophosphate (Dol-PP). The 14-sugar oligosaccharide is comprised of 2 N-acetylglucosamine, 9 mannose, and 3 glucose residues. Once assembled, the oligosaccharide is transferred en bloc to proteins and undergoes further processing in the ER and Golgi6. Once the oligosaccharide is attached to a protein, it is referred to as an N-glycan.
LLO synthesis is carried out by a series of enzymes encoded by the DPAGT1 and ALG genes and can be divided into two phases: Phase I and Phase II 6 (Figure 1).
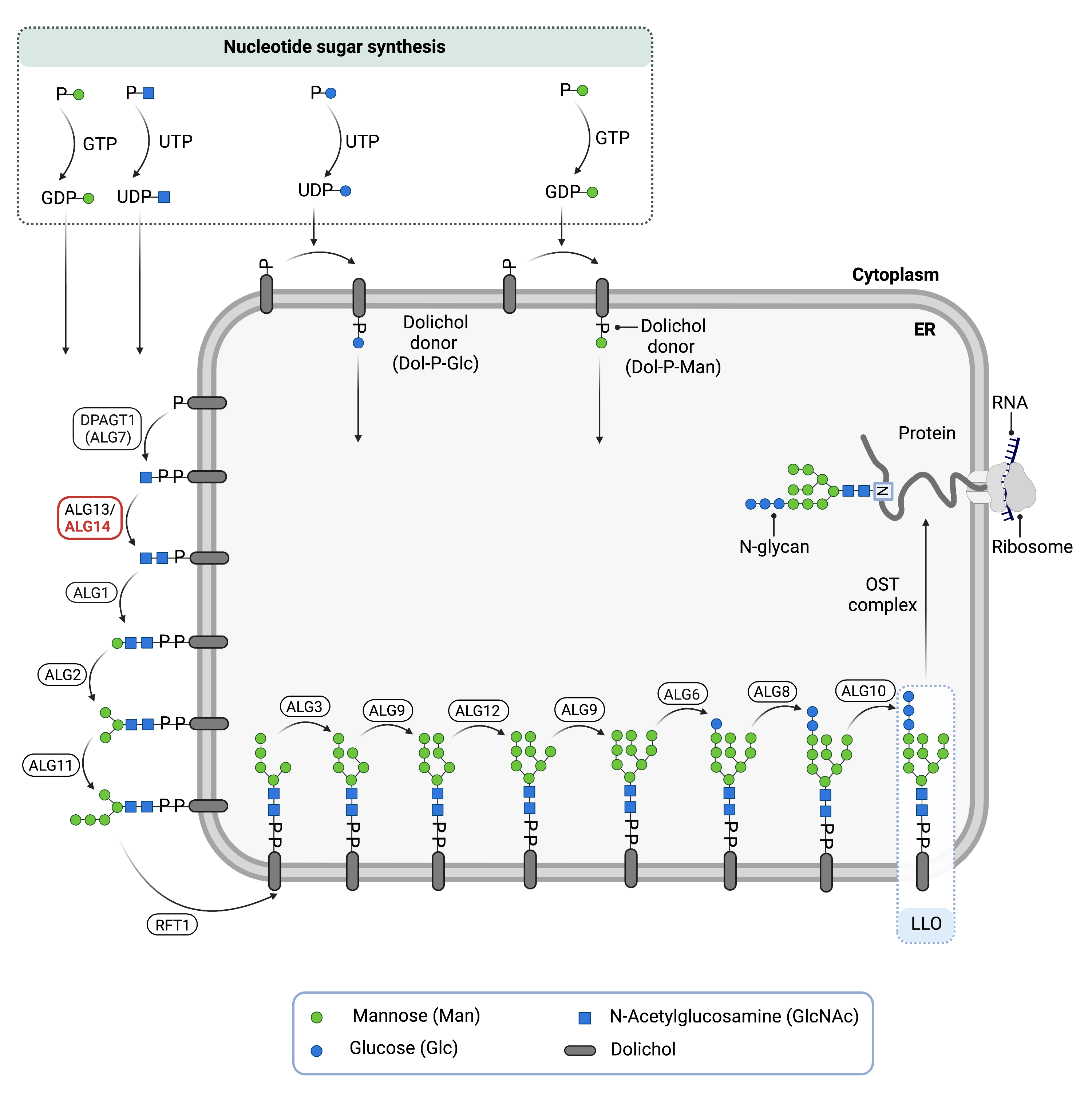
Figure 1. Role of ALG14 in Glycosylation.
ALG14 is an enzyme (ALG14 UDP-N-Acetylglucosaminyltransferase Subunit) involved in synthesizing the lipid-linked oligosaccharide (LLO) in N-glycosylation. ALG13 and ALG14 are subunits of UDP-GlcNAc transferase complex; it catalyzes the transfer of a GlcNAc residue from UDP-GlcNac to the LLO on the cytosolic side of the endoplasmic reticulum membrane.
Phase I
Phase I of LLO synthesis takes place on the cytoplasmic side of the ER. It begins with the sequential attachment of two N-Acetylglucosamine (GlcNAc2) and five mannose (Man5) residues to Dol-PP by several ER enzymes. GlcNAc and mannose are transferred from nucleotide sugars UDP-GlcNAc and GDP-mannose, which are located in the cytoplasm. UDP-GlcNAc transferase, comprised of ALG13 and ALG14 subunits, transfers the GlcNac residue from UDP-GlcNAc to Dol-PP-GlcNAc. The intermediate structure, Dol-PP-GlcNAc2Man5, is translocated from the cytoplasm into the ER lumen by the RFT1 enzyme6,8.
Phase II
Phase II of LLO synthesis takes place in the ER lumen. Once in the lumen, four mannose residues followed by three glucose (Glc3) residues are added to the intermediate structure, generating the complete the LLO, Dol-PP-GlcNAc2Man9Glc3. Mannose and glucose are transferred from glycosyl donors Dol-P mannose and Dol-P-glcuose, which are formed on the cytoplasmic side of the ER and must also be flipped across the ER membrane6.
Once assembled, the oligosaccharide is transferred en bloc from Dol-PP to asparagine residues of newly synthesized protein via the enzyme oligosaccharyltransferase (OST), resulting in N-glycosylation of the protein6. The activity of OST is highly specific for the completely assembled 14-sugar N-glycan, Glc3Man9GlcNAc2.
Disease Mechanism
Mutations in the ALG14 gene lead to the production of an abnormal protein, that is unable to function as the membrane-bound subunit of UDP-GlcNAc transferase. As a result, LLO synthesis is incomplete and the transfer of its glycan component onto proteins is impaired and N-glycoproteins are under-glycosylated (hypoglycosylation).
Defective glycosylation can also cause CMS which impairs signal transmission at the neuromuscular synapse. Silencing of ALG14 via RNAi results in reduced expression of muscle acetylcholine receptors at the motor endplate, which is suggested as the primary disease mechanism9.
Mutations
The ALG14 gene is found on Chromosome 1 (1p21.3). Homozygous splicing variant c.420+9delTAAG and truncation variant p.Arg104* produce a mild clinical phenotype9,10. Missense mutations (p.Asp74Asn, p.Val141Gly, and p.Arg109Gln) have been reported to cause a more severe clinical phenotype1,3.
Signs & Symptoms
Clinical Presentation
Individuals with ALG14-CDG typically have either a severe, multisystemic presentation or a milder presentation that primarily involves muscle weakness (ALG14-CMS).
ALG14-CMS
Individuals with ALG14-CDG typically develop signs and symptoms during infancy or prenatally. ALG14-CDG is primarily characterized by congenital myasthenic syndrome and joint contractures. The characteristic clinical presentations of ALG14-CDG include1:
- Neurological symptoms – developmental delay, infantile spasms, epilepsy, low muscle tone (hypotonia), muscle weakness (myopathy) and chronic immune disorder myasthenia.
- Dysmorphic features – contractures at knees, elbows, and ankles, turricephalus, scaphocephalus, microgathia, narrow thorax, wide set mamillae, arachnodactylia, pesesquinus, and facial anomalies such as blepharophimosis.
Brain MRI results show delayed myelination, as well as frontoparietal atrophy, ventriculomegaly, and white matter volume loss.
ALG14-CMS
Individuals with ALG14-CMS typically present with symptoms in childhood which is primarily characterized by muscle weakness. Patients with ALG14-CMS do not display the other multi-system symptoms associated with ALG14-CDG. The characteristic clinical presentations of ALG14-CMS include:
- Neurological symptoms – severely affected proximal and distal limb muscle weakness, mildly affected facial, ocular, bulbar, and respiratory muscles
Cases of two siblings with severe CMS has been reported4.
Biochemical Abnormalities
No biochemical abnormalities are reported.
Classification
ALG14-CDG is classified as a disorder of N-linked protein glycosylation.
Under the former CDG classification system, ALG14-CDG is classified as a Type 1 CDG which arise due to defects in the production of lipid-linked oligosaccharides or their transfer to proteins.
Diagnosis
Although diagnosis of ALG14-CDG may be suspected based on presentation of symptoms and a detailed patient history, direct molecular genetic testing is the only definitive diagnostic test. For the majority of CDG-I types, a type 1 pattern is observed by transferrin analysis. However, ALG14-CDG patients show normal transferrin patterns by transferrin isoelectric focusing (TIEF), therefore it is not a reliable diagnostic test for ALG14-CDG. Direct molecular genetic testing is required for diagnosis1. Specialized testing on muscle and nerves is typically carried out when ALG14-CMS is suspected.
Transferrin Analysis
Individuals with ALG14-CDG show normal patterns by transferrin analysis, therefore it is not reliable for diagnosis and other biomarkers are needed.
Muscle Biopsy
Muscle biopsies of patients with ALG14-CMS showed myopathic and dystrophic alterations, signs of disease affecting the T-tubule3. Tubular aggregates were not observed in muscle biopsies of patients with ALG14-CMS, as they have in patients with DPAGT1-CMS9.
Repetitive Nerve Stimulation
A repetitive nerve stimulation test may reveal an abnormal decrease in compound muscle action potential in patients. RNS testing is useful for early diagnosis, as results can point to pathogenesis due to AChR deficiency, which is thought to be caused by mutations to ALG144,9.
Biomarkers
No biomarkers have been reported for ALG14-CDG.
Prognosis
Prognosis of ALG14-CDG may vary depending on the severity of an individual’s symptoms. Severely affected patients may die within the first year of life from complications due to myasthenic and myopathic features as well as cerebral atrophy and epilepsy1. Mild cases have been reported where the onset of myasthenic symptoms began at age 7 and 40 in sisters, who also did not show symptoms of epilepsy or cranial abnormalities2.
Two siblings have been reported with severe CMS, presenting with hypotonia, multiple joint contractures, and swallowing and respiratory problems leading to nasal feeding and tracheotomy. Treatment with pyridostigmine improved respiratory problems and epilepsy was treated with anti-epileptic medication. Currently they are 5 and 2 years old, making them the oldest reported patients with severe CMS due ALG14 mutations4.
Management
Management of symptoms may include combinations of physical therapy, occupational therapy, speech or vision therapy and palliative measures.
Mild cases of ALG14-CDG have reported that patients have seen long-term benefits from treatment with cholinesterase inhibitors, showing improvement in muscle related symptoms due to CMS. Pyridostigmine also showed temporary improvement in some patients1.
Therapies
Treatment for other symptoms is focused on management of symptoms and prevention of complications.
Research Models
Several ALG14 research models have been generated including yeast, fly and mouse model organisms.
Yeast (S. cerevisiae)
Alg14 mutants have reduced function and show slow growth, defective protein glycosylation and accumulate LLOs with one GlcNAc residue (YeastGenome). The mechanisms by which Alg14 recruits catalytic partners Alg13 and Alg7 to the ER membrane to initiate N-glycosylation has been delineated in yeast11,12.
Fly (D. melanogaster)
Drosophila Alg14 (CG6308, FBgn0030645) is orthologous to ALG14 and predicted to be part of UDP-N-acetylglucosamine transferase complex (FlyBase).
Mouse (M. musculus)
Homozygous Alg14-/- knockout
Homozygous whole body Alg14 knockout mice have been generated from the mouse line Alg14tm2b(EUCOMM)Hmgu. Mutants live from E9.5 to early adulthood. Mutants living to E9.5 phenotypically present with embryonic lethality prior to organogenesis. Mutants living to early adulthood present with preweaning lethality, and complete penetrance(IMPC).
Alg14 conditional-ready floxed knockout ES cell line
Embryonic Stem (ES) cell line Alg14tm2a(EUCOMM)Hmgu is a targeted knockout/null mutation of Alg14. A “conditional-ready” allele can be created by flp recombinase expression in mice carrying this allele, and cre expression results in a knockout mouse. If cre is expressed without flp expression, a reporter knockout mouse can be generated (MGI).
Alg14 knockout ES cell line
Embryonic Stem (ES) cell line Alg14tm2e(EUCOMM)Hmgu are targeted knockout/null mutations of Alg14 (MGI).
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including ALG14-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Publications
ALG14-CDG Scientific Articles on PubMed
Additional Resources
References
- Schorling, D. C. et al. Early and lethal neurodegeneration with myasthenic and myopathic features. Neurology 89, (2017).
- Cossins, J. et al. Congenital myasthenic syndromes due to mutations in ALG2 and ALG14. Brain 136, (2013).
- Palombo, F. et al. A novel ALG14 missense variant in an alive child with myopathy, epilepsy, and progressive cerebral atrophy. American Journal of Medical Genetics Part A 185, (2021).
- Katata, Y. et al. The longest reported sibling survivors of a severe form of congenital myasthenic syndrome with the ALG14 pathogenic variant. Am J Med Genet A 188, 1293–1298 (2022).
- Gao, X.-D., Tachikawa, H., Sato, T., Jigami, Y. & Dean, N. Alg14 Recruits Alg13 to the Cytoplasmic Face of the Endoplasmic Reticulum to Form a Novel Bipartite UDP-N-acetylglucosamine Transferase Required for the Second Step of N-Linked Glycosylation. Journal of Biological Chemistry 280, (2005).
- Bieberich, E. Synthesis, Processing, and Function of N-glycans in N-glycoproteins. Adv Neurobiol 47-70 (2014).
- Harada, Y., Ohkawa, Y., Kizuka, Y. & Taniguchi, N. Oligosaccharyltransferase: A gatekeeper of health and tumor progression. International Journal of Molecular Sciences 20, 1-14 (2019).
- Stanley, P., Taniguchi, N. & Aebi, M. N-glycans. in Essentials of Glycobiology [Internet] (eds. Varki, A. et al.) (Cold Spring Harbor Laboratory Press, 2017).
- Cossins, J. et al. Congenital myasthenic syndromes due to mutations in ALG2 and ALG14. Brain 136, 944–956 (2013).
- Kvarnung, M. et al. Genomic screening in rare disorders: New mutations and phenotypes, highlighting ALG14 as a novel cause of severe intellectual disability. Clinical Genetics 94, 528–537 (2018).
- Gao XD, Moriyama S, Miura N, Dean N, Nishimura S. Interaction between the C termini of Alg13 and Alg14 mediates formation of the active UDP-N-acetylglucosamine transferase complex, J Biol Chem, 283, 32534-32541 (2008).
- Mitusińska, K. et al. Structural Analysis of the Effect of Asn107Ser Mutation on Alg13 Activity and Alg13-Alg14 Complex Formation and Expanding the Phenotypic Variability of ALG13-CDG. Biomolecules 12, 398 (2022).