
Lay Summary
Congenital disorders of glycosylation (CDG) are large group of genetic disorders that arise from defects in glycosylation – the process of creating, modifying and attaching sugar chains (glycans) to proteins and lipids. Most CDG types may be classified into four groups based on the specific glycosylation pathway(s) affected, which include: disorders of (i) N-linked protein glycosylation, (ii) O-linked protein glycosylation, (iii) GPI anchor biosynthesis and lipid glycosylation and (iv) multiple glycosylation pathways. However, some CDG arise from defects in removing glycans from proteins, a process that is called deglycosylation. These CDG are classified as congenital disorders of deglycosylation (CDDG). Currently, two disorders of deglycosylation have been identified, NGLY1-CDDG and MAN2C1-CDDG, which are caused by defects in the removal of glycans from proteins and their subsequent breakdown. These processes are important for helping cells remove damaged or misfolded proteins and recycling sugars. However, it is not well understood yet by researchers how defects in these processes cause symptoms of CDDG. Although the signs and symptoms vary between these CDDG, individuals can commonly display neurological defects and intellectual disabilities. Initial screening tests commonly used to screen for and help diagnose CDG cannot be used for diagnosing CDDG, with diagnosis only achieved through genetic testing. Some biomarkers have been identified for NGLY1-CDDG, which may aid in diagnosis. There are currently no approved therapies for CDDG, but various therapeutic approaches are being evaluated for NGLY1-CDDG.
Overview
Congenital disorders of glycosylation (CDG) arise from defects in protein and lipid glycoyslation. Glycosylation can be divided into different pathways, depending on whether the glycans are being added to a protein or lipid and how these glycans are attached, such as N-linked protein glycosylation, O-linked protein glycosylation, glycosylphosphatidylinositol (GPI) anchor biosynthesis, and lipid glycosylation2. CDG can then be classified into groups based on which of these glycosylation pathways are affected1. However, to date, only two disorders, have been linked to abnormal deglycosylation and glycan degradation, which is the process of removing glycans from proteins or lipids and breaking down glycans. These disorders are classified as congenital disorders of deglycosylation (CCDG) and include NGLY1-CDDG and MAN2C1-CDDG Both of these CDDG affect the deglycosylation and degradation of N-glycoproteins and the breakdown of N-glycans4.
N-Glycoprotein Degradation
N-linked protein glycosylation (or N-glycosylation) involves the attachment of a 14-sugar oligosaccharide (Glc3Man9GlcNAc2) to the nitrogen atom of asparagine (Asn) residues on a protein as it is being synthesized in the endoplasmic reticulum (ER) lumen. The oligosaccharide is built on the lipid carrier dolichol, forming a lipid-linked oligosaccharide (LLO). The oligosaccharide is transferred from the LLO onto a newly-synthesised protein—where the protein with attached glycan (N-glycan) is referred to as an N-glycoprotein. Next, enzymes remove two glucose residues from the N-glycan (forming GlcMan9GlcNAc2), where the protein then associates with chaperone proteins, calnexin and calreticulin, to undergo folding6. The final glucose is removed from the N-glycan, allowing the N-glycoprotein to be transported to the Golgi for further processing or to its final destination in the cell7. If the protein is not folded properly, glucose can be re-added to the N-glycan by the UGGT enzyme, and the misfolded N-glycoprotein can be passed back to chaperone proteins for refolding. However, if N-glycoprotein refolding fails, the glycoprotein can eventually be broken down through lysosomal degradation or non-lysosomal and endoplasmic reticulum associated degradation (ERAD), where both the protein and glycan are degraded (Figure 1). In addition to misfolded proteins, properly folded N-glycoproteins are also degraded as part of healthy cellular functioning8. Faulty N-glycoprotein degradation can lead to disease, such as lysosomal storage diseases and CDDG4,9,10.
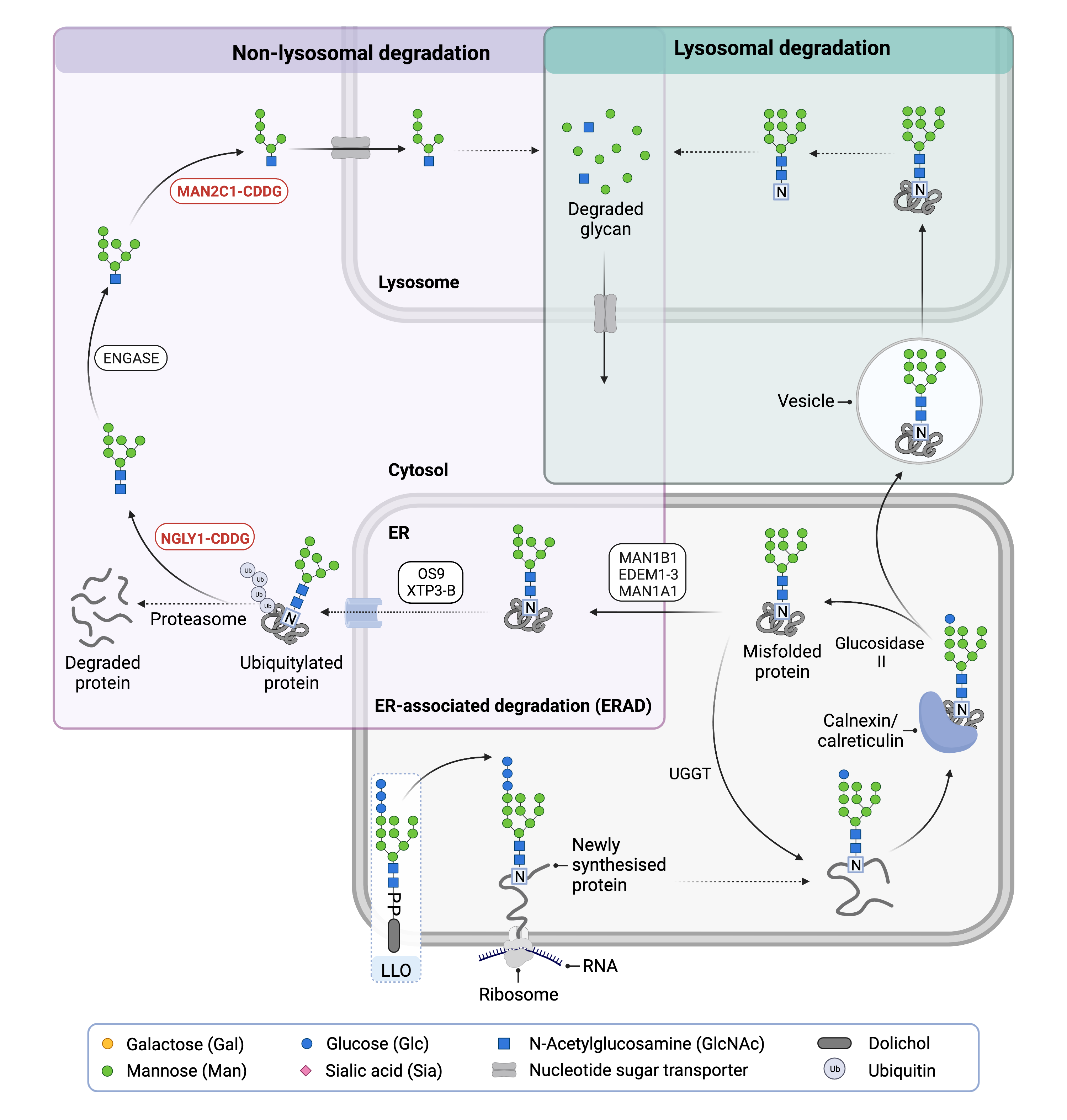
Figure 1. Overview of lysosomal and non-lysosomal degradation of N-glycoproteins.
In lysosomal degradation, glycoproteins are transported to the lysosome, where enzymes break down the glycoprotein and release the amino acids and monosaccharides back into the cytosol. In non-lysosomal degradation, misfolded N-glycoproteins in the ER are degraded in the cytosol through ERAD. The protein is degraded through the proteasome, while the glycan is degraded in the cytosol and the lysosome.
Lysosomal Degradation
Lysosomes are compartments located in the cytosol of the cell that contain a variety of enzymes that degrade glycans and other molecules11. For lysosomal degradation, N-glycoproteins are transported to the lysosome either from the cell surface or extracellular space via endocytosis or intracellularly by other delivery processes. Enzymes, called proteases, then begin degrading the protein by breaking the bonds joining amino acids. Once most of the protein has been degraded, and only an asparagine amino acid remains attached to the N-glycan, enzymes called glycosidases then remove the N-glycan from asparagine and break down the glycan into monosaccharides4. The amino acids and monosaccharides are then released into the cytosol, ready to be reused for making new proteins and glycans8.
Non-Lysosomal Degradation
N-glycoproteins may also under undergo non-lysosomal degradation in the cytosol through ER-associated degradation (ERAD), a quality control process for misfolded glycoproteins12. If protein folding fails, proteins are targeted for ERAD which begins with the sequential removal of terminal mannose residues from the misfolded N-glycoproteins by mannosidases. Here, mannosidases remove or trim terminal mannose residues from the N-glycan of a misfolded N-glycoprotein, forming Man7GlcNAc2. The removal of the mannose allows ER degradation-enhancing α-mannosidase-like proteins (EDEMs) to remove the trimmed glycan from calnexin and calreticulin6. Next ER proteins (OS-9 and XTP3-B) deliver the trimmed N-glycoprotein to a protein complex located at the ER membrane6,13. Here, the misfolded glycoprotein is moved from the ER to the cytosol, and ubiquitin molecules are attached to the protein7.
The ubiquitinated N-glycoprotein then undergoes degradation in the cytosol. The glycan is removed from the protein by a deglycosylating enzyme (NGLY1), where the unattached glycan is now referred to as a ‘free N-glycan’ or ‘free oligosaccharide’4. The protein is degraded through the proteasome, while the free N-glycan is further broken down by enzymes ENGASE and MAN2C14,7. ENGASE removes the terminal GlcNAc residue and MAN2C1 removes mannose residues from the free N-glycan, generating Man5GlcNAc, which is then transported to the lysosome for further degradation of the remaining glycan chain4.
Disorders of Deglycosylation
To date, NGLY1-CDDG and MAN2C1-CDDG are the only disorders of deglycosylation to be identified.
NGLY1-CDDG
NGLY1-CDDG, also commonly referred to as NGLY1 deficiency, was the first disorder of deglycosylation to be identified. NGLY1-CDDG is caused by mutations in the NGLY1 gene which encodes for N-glycanase I (NGLY1), an enzyme that catalyzes the removal of N-linked glycans (N-glycans) from proteins. NGLY1 is important for the degradation and recycling of misfolded N-glyocproteins via ERAD and may additional roles in various cellular processes such as lipid metabolism and mitochondrial function14,15. NGLY1-CDDG is primarily characterized by a lack of absence of tears (alacrimia), reduced muscle tone (hypotonia), neurodevelopmental delay, seizures, abnormal involuntary movements and liver abnormalities3.
MAN2C1-CDDG
MAN2C1-CDDG was recently identified as a disorder of deglycosylation. It is caused by mutations in the MAN2C1 gene which encodes alpha-mannosidase (MAN2C1), an enzyme found in the cytoplasm that removes mannose from free N-glycans during ERAD of glycoproteins4. Of the 6 cases documented to date, individuals with MAN2C1-CDDG present with abnormal facial features, variable intellectual disability and abnormalities in several brain structures16.
Diagnosis
As with many CDG, individuals with disorders of deglycosylation require genetic testing for definitive diagnoses10,17. Traditional tests used in CDG diagnosis are not reliable methods for diagnosing NGLY1-CDDG, such as apolipoprotein C-III analysis, transferrin analysis, and total glycan analysis and has not yet been investigated for MAN2C1-CDG9,17. Several potential biomarkers for NGLY1-CDDG have been identified which may aid in the diagnosis of patients18–20.
References
- Lipiński, P. & Tylki-Szymańska, A. Congenital Disorders of Glycosylation: What Clinicians Need to Know? Frontiers in Pediatrics vol. 9 (2021).
- Ondruskova, N., Cechova, A., Hansikova, H., Honzik, T. & Jaeken, J. Congenital disorders of glycosylation: Still “hot” in 2020. Biochimica et Biophysica Acta (BBA) - General Subjects 1865, (2021).
- Lam, C. et al. Prospective phenotyping of NGLY1-CDDG, the first congenital disorder of deglycosylation. Genetics in Medicine 19, (2017).
- Suzuki, T. Catabolism of N-glycoproteins in mammalian cells: Molecular mechanisms and genetic disorders related to the processes. Molecular Aspects of Medicine vol. 51 (2016).
- Reily, C., Stewart, T. J., Renfrow, M. B. & Novak, J. Glycosylation in health and disease. Nature Reviews Nephrology 15, (2019).
- Gariballa, N. & Ali, B. R. Endoplasmic Reticulum Associated Protein Degradation (ERAD) in the Pathology of Diseases Related to TGFβ Signaling Pathway: Future Therapeutic Perspectives. Frontiers in Molecular Biosciences vol. 7 (2020).
- Zhang, J., Wu, J., Liu, L. & Li, J. The Crucial Role of Demannosylating Asparagine-Linked Glycans in ERADicating Misfolded Glycoproteins in the Endoplasmic Reticulum. Frontiers in Plant Science vol. 11 (2021).
- Winchester, B. Lysosomal metabolism of glycoproteins. Glycobiology vol. 15 (2005).
- Lam, C., Wolfe, L., Need, A., Shashi, V. & Enns, G. NGLY1-Related Congenital Disorder of Deglycosylation. GeneReviews® (1993).
- Maia, N. et al. Impaired catabolism of free oligosaccharides due to MAN2C1 variants causes a neurodevelopmental disorder. The American Journal of Human Genetics (2022) doi:10.1016/j.ajhg.2021.12.010.
- Freeze, H. H., Kinoshita, T. & Schnaar, R. L. Genetic Disorders of Glycan Degradation. Essentials of Glycobiology (2015).
- Roth, J. & Zuber, C. Quality control of glycoprotein folding and ERAD: the role of N-glycan handling, EDEM1 and OS-9. Histochemistry and Cell Biology vol. 147 (2017).
- Eura, Y., Miyata, T. & Kokame, K. Derlin-3 is required for changes in erad complex formation under ER stress. International Journal of Molecular Sciences 21, (2020).
- Fujihira, H., Asahina, M. & Suzuki, T. Physiological importance of NGLY1, as revealed by rodent model analyses. The Journal of Biochemistry (2021) doi:10.1093/jb/mvab101.
- Galeone, A. et al. Tissue-specific regulation of BMP signaling by Drosophila N-glycanase. eLife 6, (2017).
- Wang, L. & Suzuki, T. Dual functions for cytosolic α-mannosidase (Man2C1) its down-regulation causes mitochondria-dependent apoptosis independently of its α-mannosidase activity. Journal of Biological Chemistry 288, (2013).
- Enns, G. M. et al. Mutations in NGLY1 cause an inherited disorder of the endoplasmic reticulum-associated degradation pathway. Genetics in medicine : official journal of the American College of Medical Genetics 16, (2014).
- Hall, P. L. et al. Urine oligosaccharide screening by MALDI-TOF for the identification of NGLY1 deficiency. Molecular Genetics and Metabolism 124, (2018).
- Haijes, H. A. et al. Aspartylglycosamine is a biomarker for NGLY1-CDDG, a congenital disorder of deglycosylation. Molecular Genetics and Metabolism 127, (2019).
- Mueller, W. F. et al. GlcNAc-Asn is a biomarker for NGLY1 deficiency. The Journal of Biochemistry (2021) doi:10.1093/jb/mvab111.