
Lay Summary
Sugar chains are attached to fats (lipids) in the body through two major pathways: the synthesis of glycosylphosphatidylinositol (GPI) anchors and lipid glycosylation. Many different proteins are attached to GPI molecules at the cell membrane, which effectively “anchor” the protein to the surface of the cell; these proteins are called GPI-anchored proteins. In the context of congenital disorders of glycosylation (CDG), lipid glycosylation refers specifically to the generation of glycosphingolipids (GSLs)1. GSLs are an important component of the cell membrane and are particularly abundant in brain cells. CDG that arise due to defects in either of these pathways are classified as disorders of GPI anchor biosynthesis and lipid glycosylation. To date, 24 CDG involving GPI anchor biosynthesis have been identified but only three CDG involving GSL biosynthesis are currently known. Due to the important roles that GPI-anchored proteins and GSLs play in many different biological pathways, multiple systems are usually affected when their biosynthesis is impaired. Although symptoms, severity and age of onset vary in CDG involving GPI anchor biosynthesis and GSL biosynthesis, some common signs include developmental delays, neurologic disease, seizures, poor muscle tone, abnormal facial features, problems with blood clotting, and elevated alkaline phosphatase levels; issues with the heart, liver, and kidneys are also common1. Genetic testing is typically required for definitive diagnosis.
Overview
Glycosylation plays a key role in two major lipid-based pathways: the biosynthesis of glycosylphosphatidylinositol (GPI) anchors and glycosphingolipids (GSLs)1. GPI anchors are an important mechanism of attachment for proteins to the cell surface, and these proteins are known as GPI-anchored proteins2,3. GSLs are present in all the plasma membrane of all human cells but are abundant in nervous tissue and are the major glycan of the brain1,4. GPI-anchored proteins and GSLs are not evenly distributed throughout the plasma membrane, but instead concentrate into small, transiently formed microdomains on the cell surface known as “lipid rafts”5–8. Self-assembly into lipid rafts can help bring interacting proteins closer together to initiate intracellular signaling. Like other classes of CDG, disorders of GSL and GPI-anchored protein biosynthesis are extremely rare, can manifest very different symptoms, and usually require next generation sequencing (NGS) for successful diagnosis.
GPI-Anchored Protein Biosynthesis
GPI is a type of glycosylated lipid (glycolipid) that attaches proteins to the outer plasma membrane surface of cells. These GPI-anchored proteins self-associate into transient lipid rafts in the plasma membrane, and this process may facilitate their role in embryogenesis, neurogenesis, immunity, and fertilization. The protein component of GPI-anchored proteins may also be cleaved and released from the cell surface by the action of GPIases9.
The core structure of GPI consists of phosphatidylinositol (PI), glucosamine (GlcN), three mannose sugars (Man)3, and phosphoethanolamine (EtN-P) connected to each other in that sequence (Figure 1)9–11. GPI-anchored proteins are attached to the GPI by forming a bond between the EtN-P group of the GPI core and the C-terminus of newly synthesized proteins in the ER lumen.
GPI-anchored protein biosynthesis can be broken into three major steps:
- GPI anchor synthesis
- Protein attachment to the GPI anchor
- Lipid and glycan remodelling and transport of the GPI-anchor protein
The majority of this process occurs in association with the endoplasmic reticulum (ER), but modifications to the lipid component occur primarily in the Golgi before the GPI-anchored proteins are transported to the cell surface.
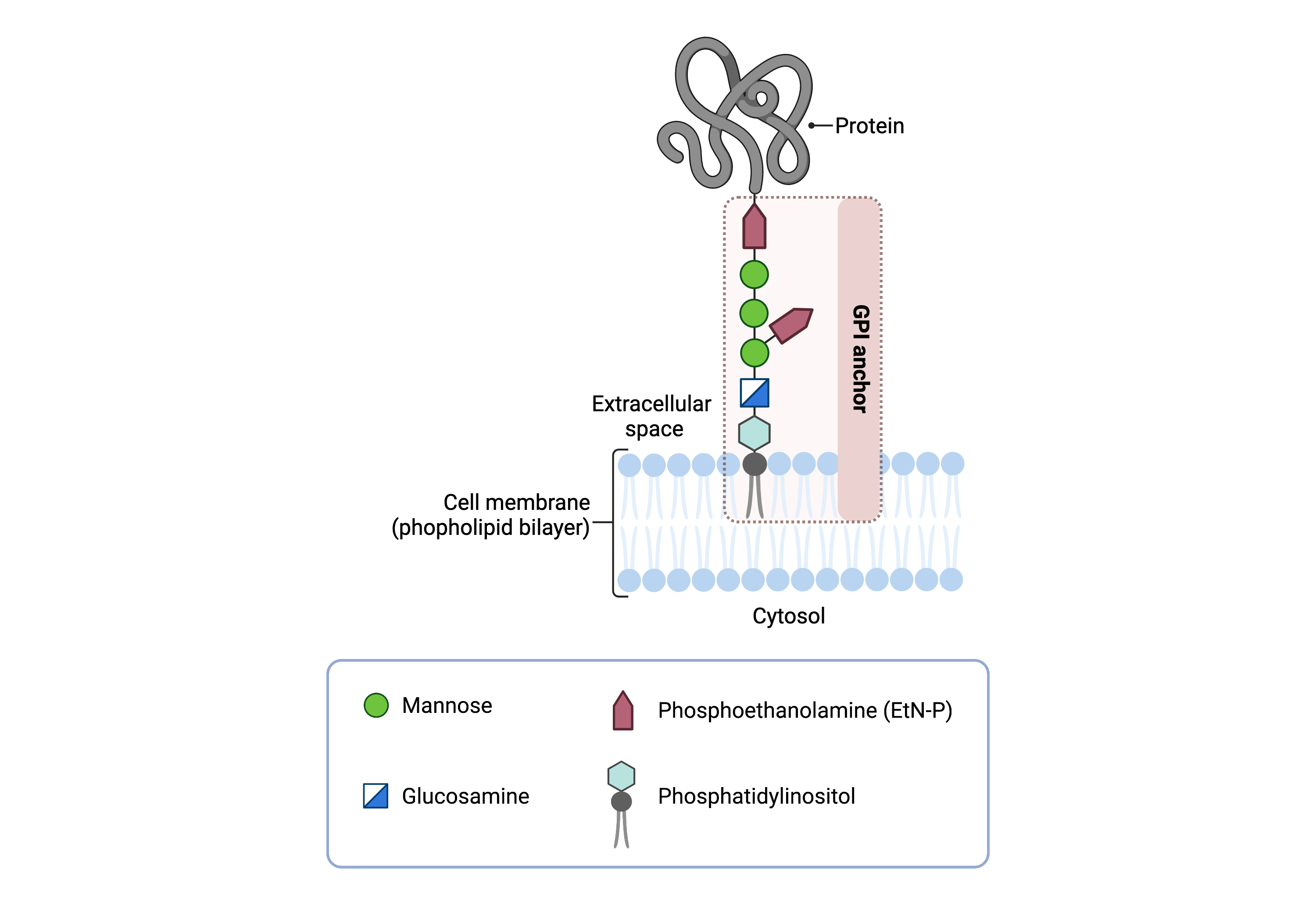
Figure 1: Overview of the GPI core structure.
The GPI core, PI-GlcN-Man3-EtN-P, contains phosphatidylinositol (PI), glucosamine, mannose, and phosphoethanolamine (EtN-P); it is attached to a protein to form a GPI-anchored protein.
GPI Anchor Synthesis
The GPI anchor core, PI-GlcN-Man3-EtN-P, consists of a glycan, GlcN-Man3-EtN-P, attached to phosphatidyl inositol (PI)7–9,12. PI is a membrane-bound glycerophospholipid with a glycerol backbone connected to two fatty acid lipid tails and an inositol sugar. Phosphatidylinositol glycan anchor biosynthesis (PIG) enzymes are responsible for building the glycan portion of the GPI anchor. The glycan chain is built off the inositol subunit of the PI, with sugars being added sequentially through the action of PIG glycosyltransferase enzymes (Figure 2). These enzymes catalyze the transfer of sugars from a glycosyl donor to molecules which “accept” sugars. As well, the lipid tails attached to the PI glycerol backbone can be modified or substituted to help ensure that the GPI-anchored protein is properly transported in the cell and ultimately localized within lipid rafts on the cell surface.
In the early steps of the GPI anchor biosynthesis, highly energetic nucleotide sugars (GDP-mannose and UDP-N-acetylglucosamine) in the cytoplasm are the glycosyl donors, but later steps of the biosynthesis occur in the ER lumen and use sugars linked to phosphorylated dolichol molecules (e.g. Dol-P-mannose) 13. Dolichol is a lipid found in the membrane of the ER. The synthesis of the GPI anchor is completed inside the ER so that it is near the newly formed proteins that will be targeted for glycosylation, and dolichol serves as a mechanism for the transport of sugars into the ER where they can be used by glycosyltransferases to finish GPI anchor construction.
GPI anchor biosynthesis begins with the transfer of the monosaccharide N-acetylglucosamine (GlcNAc) from UDP-GlcNAc to PI to generate GlcNAc-PI (Figure 2)2,7,9–11. This process requires seven proteins working together as part of a large glycosyltransferase complex called GPI GlcNAc transferase (GPI-GnT). Next, the GlcNAc sugar has its acetyl (Ac) group removed to yield GlcN-PI. GlcN-PI is then transferred from the cytoplasmic side of the ER membrane to the ER lumen.
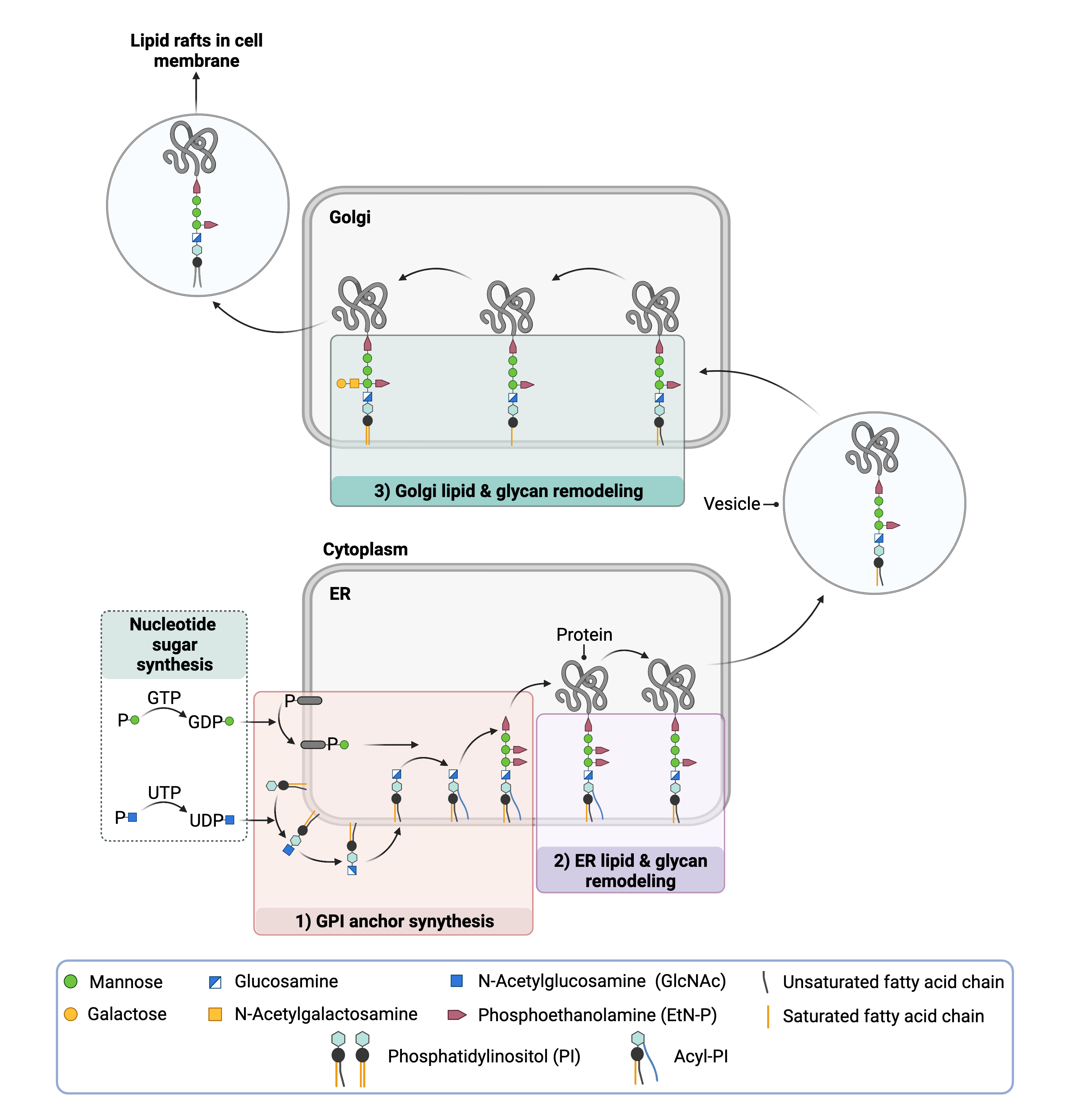
Figure 2. Overview of GPI-anchored protein biosynthesis.
Monosaccharides are transferred from activated sugars to phosphatidylinositol (PI) first in the cytosol (GDP-mannose; UDP-GlcNAc) and then in the ER (Dol-P-mannose); in the ER the lipid tails attached to PI are also modified. The GPI anchor is then transferred to a protein, forming the GPI-anchored protein, which undergoes glycan and lipid modifications in the ER and Golgi.
After being flipped into the ER lumen, the lipid component of the GPI anchor undergoes significant changes. First, a fatty acid chain is added to the inositol by an enzyme, yielding GlcN-(acyl)PI. This group is thought to facilitate later attachment of the protein to the anchor. Then, the lipid tails attached to the glycerol are exchanged with other lipids by currently unidentified enzyme(s)7,9,10,12.
The final steps of the GPI anchor synthesis involve completion of the glycan component by the sequential addition of mannose sugars and EtN-P groups. Typically, the final form of the GPI anchor has three mannose sugars each with an EtN-P group attached. In the brain, it is common for a fourth mannose sugar to be added to the glycan chain9.
Protein Attachment to the GPI Anchor
Once the GPI anchor has been synthesized, it is transferred "en bloc" to a protein with a C-terminal GPI attachment signal sequence. This amino acid sequence is variable but ensures that proteins meant to have GPI anchors are recognized by the five-protein GPI transamidase (GPI-TA) complex. This complex catalyzes the simultaneous cleavage of the signal sequence and attachment of the GPI anchor to the newly synthesized protein2,6,7,10.
Remodeling and Protein Transport
After the protein has been attached to the GPI anchor, both the glycan and lipid portion of the anchor undergo modifications (referred to as remodelling) in the ER and Golgi by post-GPI attachment to protein (PGAP) enzymes. In the ER, remodeling includes removal of the inositol-linked fatty acid and removal of one of the EtN-P attached to the glycan chain. These changes allow the GPI-anchored protein to exit the ER and be transported to the Golgi in vesicles12–15. Inositol modification is also important for the degradation of misfolded GPI-anchored proteins and subsequent modifications that occur to the lipid portion of the anchor in the Golgi. In the Golgi, the lipid portion of the GPI-anchored protein is exchanged with different lipids that better enable GPI-anchored proteins to self-assemble into lipid rafts in the cell membrane and interact with glycosphingolipids and cholesterol. Some GPI-anchored proteins undergo further modification by the addition of a N-acetlygalactosamine (GalNAc) glycan before being transported to the cell membrane.
Disorders of GPI Anchor Biosynthesis
Currently, defects in 24 of the 29 genes identified the biosynthesis of GPI-anchored proteins are known to cause CDG; this class of CDG is sometimes called inherited GPI deficiencies16. Some CDG involving GPI-anchored protein biosynthesis are also associated with hyperphosphatasia with mental retardation (HPMR) syndrome and multiple congenital anomalies-hypotonia-seizures (MCAHS) syndrome1,13,16. CDG can be caused any mutation that prevents the normal formation of GPI-anchored proteins, including errors in the synthesis of the GPI anchor, attachment of the target protein to the anchor, and subsequent lipid and glycan remodeling.
Like all CDG, disorders of GPI anchor biosynthesis vary widely in their clinical presentation but typically manifest symptoms in multiple systems. Common presentations include neurological abnormalities and developmental/cognitive delay, seizures, facial abnormalities, poor muscle tone (hypotonia), and/or clotting abnormalities. Alkaline phosphatase is synthesized as a GPI-anchored protein, and some disorders of GPI anchor biosynthesis are associated with elevated levels of alkaline phosphatase in the bloodstream (hyperphosphatasia)17. Abnormal bone, joint, and tendon structure may also be present. CDG associated with errors in GPI anchor biosynthesis display different phenotypes than CDG associated with the attachment of the anchor to the protein and later glycan and lipid remodelling16.
With the exception of PIGA-CDG, these CDG are inherited in an autosomal recessive fashion, meaning an affected individual inherits one defective copy of specific gene from each asymptomatic parent1. PIGA-CDG is X-linked recessive, meaning that males are much more likely to be affected than females; their mothers would possess one defective copy of the gene and one normal copy on their X chromosomes.
Disorders of GPI Anchor Biosynthesis that have been identified to date include (Figure 3):
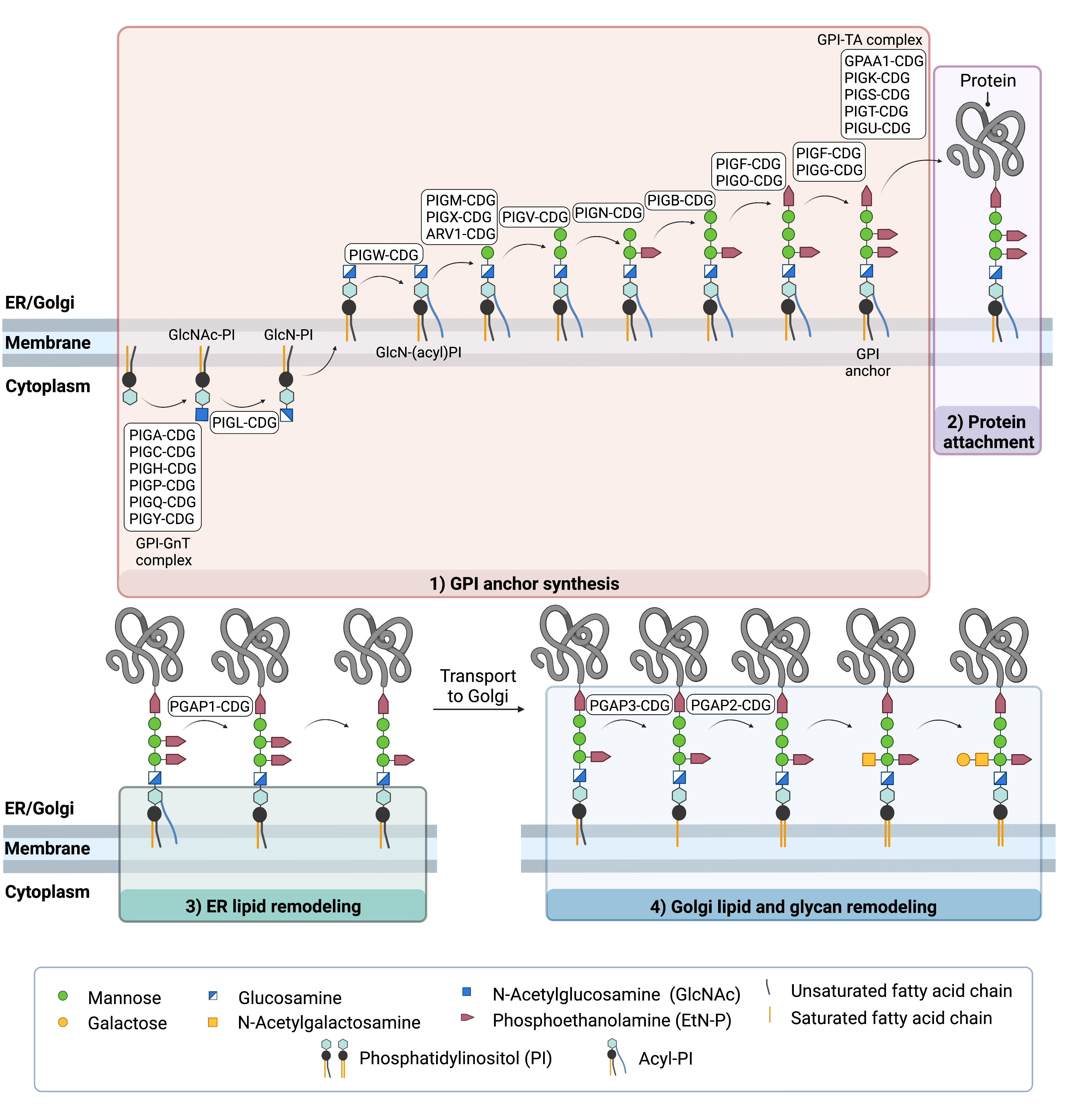
Figure 3: CDG arising from defects in GPI anchor biosynthesis.
CDG are caused by defects in GPI anchor synthesis, their attachment to proteins and lipid remodeling of GPI-anchored proteins in the ER and Golgi.
Diagnosis
Disorders of GPI anchor biosynthesis cannot be diagnosed using tests typically used in CDG screening, including analysis of transferrin and apolipoprotein-CIII abnormalities. These tests are specific for N- and O- glycosylation pathways, which are not affected in CDG involving GPI anchor biosynthesis. Instead, analysis of surface GPI-anchored proteins on blood cells can be used to diagnose some CDG types caused by defective GPI anchor biosynthesis18. As with most CDG, genetic sequencing is often necessary for definitive diagnosis of CDG due to GPI anchor biosynthesis defects.
GPI-anchored protein analysis
Cells taken from patients can be analyzed for the presence of GPI-anchored proteins on their surface using flow cytometry, which is a method of sorting cells based on a certain characteristic. One technique uses a fluorescent molecule (fluorescein-labeled proaerolysin, FLAER) that binds directly to GPI anchors in the plasma membranes of cells18.
Lipid Glycosylation
In the context of CDG, lipid glycosylation refers to the biosynthesis of a class of glycolipids called glycosphingolipids (GSLs). GSLs are an important component of the membrane of all cell types and tissues and are particularly enriched in the brain and nerve tissue19. They facilitate cell-cell recognition, cell signalling, and can also modulate the activity of nearby proteins in the cell membrane. Like GPI-anchored proteins, GSLs associate into lipid rafts in the cell membrane. The clustering of certain proteins and GSLs in raft domains might help increase the interaction between signalling molecules and their receptors, thereby facilitating messaging between cells.
The core structure of GSLs feature a glycan attached to a membrane-bound lipid ceramide (Cer) (Figure 4). Ceramides are a type of sphingolipid, made up of a fatty acid attached to the nitrogen atom of a lipid-like molecule called sphingosine. The basic structure of a GSL is a monosaccharide, usually galactose or glucose, attached to ceramide, generating galactosylceramide (GalCer) and glucosylceramide (GlcCer) respectively. GalCer and GlcCer are the simplest GSLs, and GalCer is one of the most abundant molecules in the brain19. More complex GSLs are made by extending the glycan chain on GlcCer through the action of various enzymes in the ER and the Golgi. In humans, most GSLs may be broadly classified into 5 groups based on the sugar composition of their glycan chain (Figure 4): ganglioside, globoside, isogloboside, lactoside, and neolactoside. Within these groups, GSLs can be further subclassified.
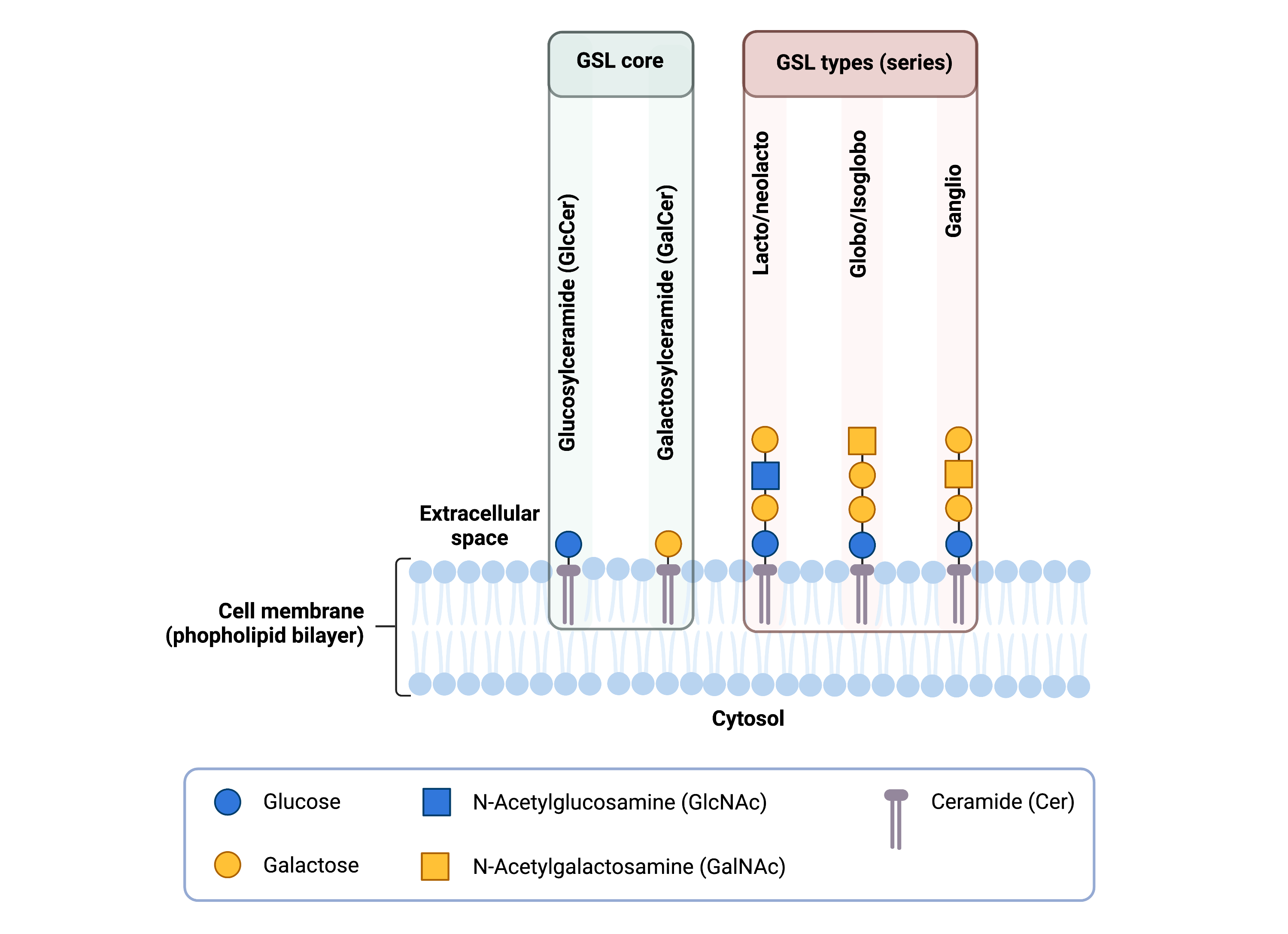
Figure 4: Overview of glycosphingolipid (GSL) structures.
The basic structure of GSLs typically include glucose or galactose attached to ceramide; GlcCer and GalCer are the simplest GSLs. Complex GSLs can be generated by elongating the glycan on GlcCer with additional monosaccharides and can be classified into 5 groups (series) based on the sugars in the glycan: lactoside, neolactoside globoside, isogloboside or ganglioside.
Glycosphingolipid Biosynthesis
Biosynthesis of complex GSLs starts with the attachment of glucose to the sphingolipid ceramide on the cytoplasmic side of the ER and the Golgi, forming GlcCer. GlcCer is then flipped into the Golgi lumen where galactose is added, forming LacCer. LacCer is used as a starting point for the synthesis of more complex GSLs, including gangliosides and globosides1,19,20. For example, gangliosides are further subdivided into three series (a, b, and c) based on the number of sialic acids subsequently added to the LacCer core (Figure 5)19. The stepwise addition of sialic acid sugars by sialyltransferases constitute significant branch points in ganglioside biosynthesis. Ultimately, the differences in function of more complex GSLs are determined by the different sugars that are subsequently added to the glycan chain and the way they are connected to each other.
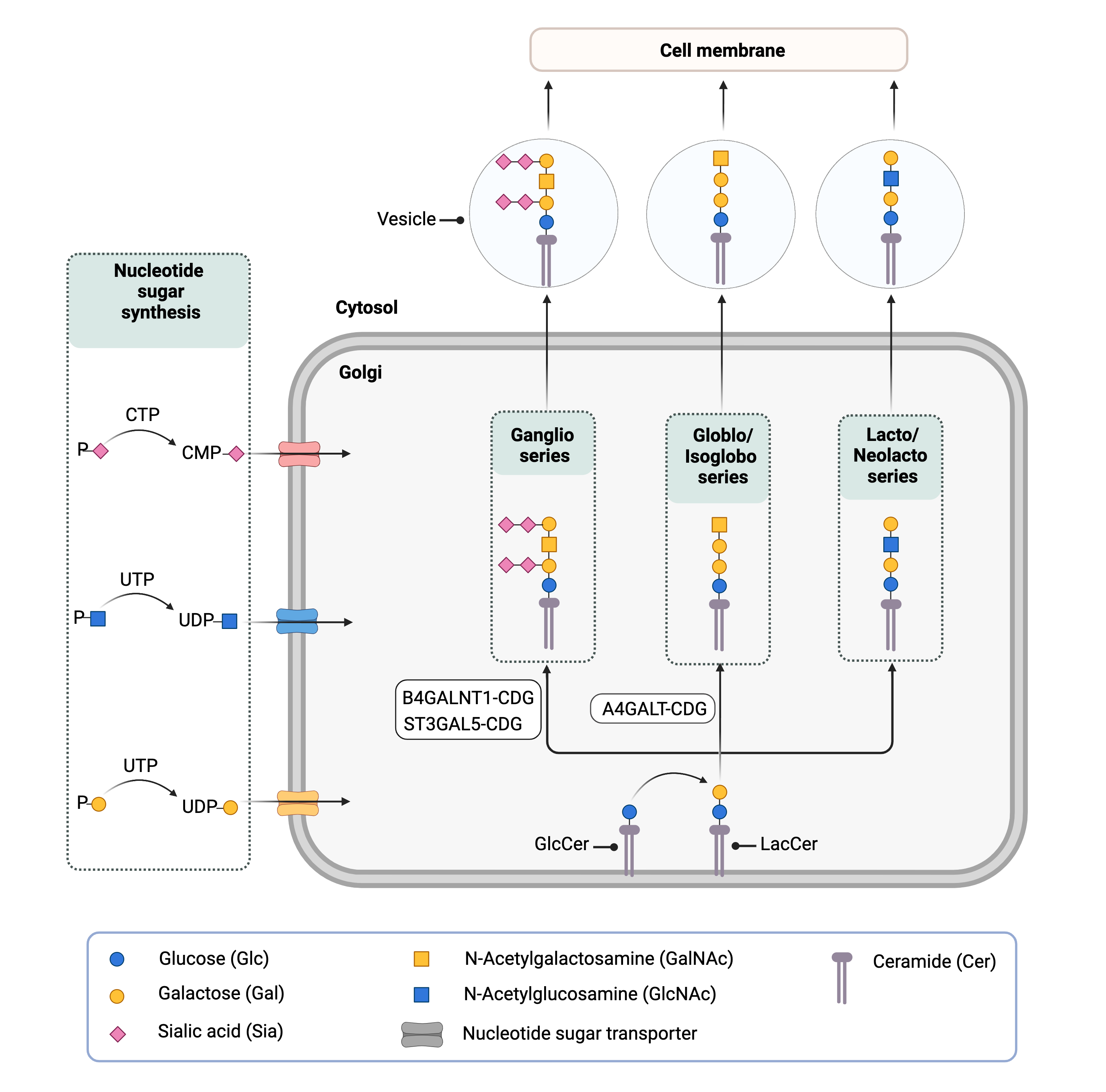
Figure 5. Overview of GSL biosynthesis and CDG caused be defects in lipid (GSL) glycosylation.
Biosynthesis of complex GSLs begins with the attachment of glucose to ceramide in the ER and Golgi, forming GlcCer. In the Golgi, galactose is added to generate LacCer; different types of complex GSLs can be generated from LacCer depending on the sugars subsequently attached to the glycan. Three CDGs caused by defects in GSL biosynthesis have been identified.
Disorders of Lipid Glycosylation
Currently, there are only three known CDG involving lipid glycosylation (Figure 5). Two involve the synthesis of gangliosides (B4GALNT1-CDG and ST3GAL5-CDG), which are a type of GSL that contain one sialic acid group and are particularly abundant in brain and nervous tissue. The third involves the synthesis of globosides (A4GALT-CDG), a type of GSL that are most abundant in red blood cells. These disorders are inherited in an autosomal recessive fashion, with affected individuals receiving one defective allele from each parent.
CDG involving ganglioside biosynthesis may cause cognitive impairment, developmental delay, abnormal involuntary movements, musculoskeletal defects, epilepsy, blindness, and abnormal skin pigmentation20–25. The only currently known CDG involving globoside biosynthesis causes a rare blood group phenotype that is associated with increased risk of blood transfusion reactions and spontaneous abortion, but affected individuals are otherwise healthy26–28.
Disorders of lipid glycosylation that have been identified to date include:
Diagnosis
Disorders of lipid glycosylation cannot be diagnosed using tests typically used in CDG screening, including analysis of transferrin and apolipoprotein CIII abnormalities. These tests are specific for N- and O- glycosylation pathways, which are not affected in CDG involving lipid glycosylation. As such, genetic sequencing is usually necessary to definitively diagnose disorders of lipid glycosylation.
References
- Ng, B. G. & Freeze, H. H. Human genetic disorders involving glycosylphosphatidylinositol (GPI) anchors and glycosphingolipids (GSL). J. Inherit. Metab. Dis. 38, 171–178 (2015).
- Ferguson, M. A. J., Hart, G. W. & Kinoshita, T. Glycosylphosphatidylinositol Anchors. in Essentials of Glycobiology (eds. Varki, A., Cummings, R. D. & Esko, J. D.) 476–478 (Cold Spring Harbor Laboratory Press, 2017). doi:10.1101/GLYCOBIOLOGY.3E.012.
- Williams, A. F. Cell-Surface Anchoring of Proteins via Glycosylphosphatidylinositol Structures. Annu. Rev. Biochem. 57, 285–320 (1988).
- Tettamanti, G., Bonali, F., Marchesini, S. & Zambotti, V. A new procedure for the extraction, purification and fractionation of brain gangliosides. Biochim. Biophys. Acta - Lipids Lipid Metab. 296, 160–170 (1973).
- Suzuki, K. G. N. et al. Transient GPI-anchored protein homodimers are units for raft organization and function. Nat. Chem. Biol. 8, 774–783 (2012).
- Zurzolo, C. & Simons, K. Glycosylphosphatidylinositol-anchored proteins: Membrane organization and transport. Biochim. Biophys. Acta - Biomembr. 1858, 632–639 (2016).
- Kinoshita, T. & Fujita, M. Thematic Review Series: Glycosylphosphatidylinositol (GPI) Anchors: Biochemistry and Cell Biology: Biosynthesis of GPI-anchored proteins: special emphasis on GPI lipid remodeling. J. Lipid Res. 57, 6 (2016).
- Mayor, S. & Riezman, H. Sorting GPI-anchored proteins. Nat. Rev. Mol. Cell Biol. 2004 52 5, 110–120 (2004).
- Liu, Y.-S. & Fujita, M. Mammalian GPI-anchor modifications and the enzymes involved. Biochem. Soc. Trans. 48, 1129–1138 (2020).
- Ikezawa, H. Glycosylphosphatidylinositol (GPI)-Anchored Proteins. Biol. Pharm. Bull. 25, 409–417 (2002).
- Englund, P. T. The Structure and Biosynthesis of Glycosyl Phosphatidylinositol Protein Anchors. Annu. Rev. Biochem. 62, 121–138 (1993).
- Fujita, M. & Kinoshita, T. Structural remodeling of GPI anchors during biosynthesis and after attachment to proteins. FEBS Lett. 584, 1670–1677 (2010).
- Maeda, Y. & Kinoshita, T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Prog. Lipid Res. 50, 411–424 (2011).
- Fujita, M. et al. Sorting of GPI-anchored proteins into ER exit sites by p24 proteins is dependent on remodeled GPI. J. Cell Biol. 194, 61–75 (2011).
- Loizides-Mangold, U., David, F. P. A., Nesatyy, V. J., Kinoshita, T. & Riezman, H. Glycosylphosphatidylinositol anchors regulate glycosphingolipid levels [S]. J. Lipid Res. 53, 1522–1534 (2012).
- Carmody, L. C. et al. Significantly different clinical phenotypes associated with mutations in synthesis and transamidase+remodeling glycosylphosphatidylinositol (GPI)-anchor biosynthesis genes. Orphanet J. Rare Dis. 2020 151 15, 1–13 (2020).
- Bruneel, A., Cholet, S., Tran, N. T., Mai, T. D. & Fenaille, F. CDG biochemical screening: Where do we stand? Biochim. Biophys. Acta - Gen. Subj. 1864, (2020).
- Knaus, A. et al. Characterization of glycosylphosphatidylinositol biosynthesis defects by clinical features, flow cytometry, and automated image analysis. Genome Med. 2018 101 10, 1–13 (2018).
- Schnaar, R. L. & Kinoshita, T. Glycosphingolipids. in Essentials of Glycobiology (eds. Varki, A., Cummings, R. D. & Esko, J. D. et al.) vol. 10 1–99 (Cold Spring Harbor Laboratory Press, 2017).
- Boccuto, L. et al. A mutation in a ganglioside biosynthetic enzyme, ST3GAL5, results in salt & pepper syndrome, a neurocutaneous disorder with altered glycolipid and glycoprotein glycosylation. Hum. Mol. Genet. 23, 418–433 (2014).
- Simpson, M. A. et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat. Genet. 2004 3611 36, 1225–1229 (2004).
- Harlalka, G. V. et al. Mutations in B4GALNT1 (GM2 synthase) underlie a new disorder of ganglioside biosynthesis. Brain 136, 3618–3624 (2013).
- Boukhris, A. et al. Alteration of Ganglioside Biosynthesis Responsible for Complex Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 93, 118 (2013).
- Wakil, S. M. et al. Novel B4GALNT1 mutations in a complicated form of hereditary spastic paraplegia. Clin. Genet. 86, 500–501 (2014).
- Khamirani, H. J. et al. Phenotype of ST3GAL3 deficient patients: A case and review of the literature. Eur. J. Med. Genet. 64, (2021).
- Lai, Y.-J. et al. A systematic study of single-nucleotide polymorphisms in the A4GALT gene suggests a molecular genetic basis for the P1/P2 blood groups. Transfusion 54, 3222–3231 (2014).
- Hellberg, Å., Schmidt-Melbye, A.-C., Reid, M. E. & Olsson, M. L. Expression of a novel missense mutation found in the A4GALT gene of Amish individuals with the p phenotype. Transfusion 48, 479–487 (2008).
- Thuresson, B., Westman, J. S. & Olsson, M. L. Identification of a novel A4GALT exon reveals the genetic basis of the P1/P2 histo-blood groups. Blood 117, 678–687 (2011).