
Lay Summary
PGM1-CDG, formerly known as CDG-It or Glycogen Storage Disease type XIV, is a rare inherited condition that affects many systems in the body. It is a relatively common congenital disorder of glycosylation (CDG) with more than 54 patients reported in the medical literature to-date. PGM1-CDG is typically classified as a disorder of multiple glycosylation pathways and is more specifically a disorder of monosaccharide interconversion. PGM1-CDG is caused when an individual has mutations in both copies of the PGM1 gene, which provides instructions for making an enzyme that is responsible for maintaining the balance between two different forms of the sugar glucose. Mutations in the PGM1 gene cause some proteins to have incomplete or absent sugar chains and can impact the maintenance of normal sugar levels in the body. Symptoms of PGM1-CDG usually begin at infancy and affect multiple systems. The most common symptoms are growth delay, facial malformations that may include a cleft palate and/or uvula, low blood sugar, muscle weakness and difficulties with exercise, abnormal heart function, and liver disease. Neurological symptoms may also be present and include seizures and mild to moderate intellectual impairment. Several screening tests are available for PGM1-CDG, but a definitive diagnosis is achieved through genetic testing. PGM1-CDG is one of the few CDG that has a known treatment (galactose supplementation), and early diagnosis and treatment is critical for improving patient outcomes.
Overview
Phosphoglucomutase 1 congenital disorder of glycosylation (PGM1-CDG) is a rare autosomal recessive genetic disorder that arises from defects in the PGM1 (phosphoglucomutase 1) gene1,2. The PGM1 gene encodes an enzyme (PGM1) responsible for interconverting and maintaining the balance between glucose-6-phosphate (G6P) and glucose-1-phosphate (G1P)3. Glucose-6-phosphate is made as the first intermediate in the conversion of sugar into energy in our cells, whereas glucose-1-phosphate is a precursor for UDP-glucose, used to build glycogen and store energy in the body. By maintaining the balance between glucose-6-phosphate and glucose-1-phosphate, PGM1 helps ensure that the changing energy needs in our body are accommodated.
Besides its role in glycogen synthesis, UDP-glucose is an activated sugar (sugar donor) used in the construction of other glucose-containing sugar chains (glycans). It is also used in the synthesis of other activated sugars, such as dolichol-phosphate-glucose (Dol-P-glucose), which is the donor of glucose sugars inside the endoplasmic reticulum (ER), and UDP-galactose, used to donate the sugar galactose onto other molecules. These sugar donors are crucial for N-glycosylation of proteins, and deficiency in PGM1 results in insufficient and incomplete N-glycosylation of glycoproteins.
The first patient with a deficiency in phosphoglucomutase was reported in 1963, with an additional patient reported in 1988; in 2009 this deficiency was categorized as a glycogen storage disorder4–6. Further analysis of patients with PGM1 deficiency revealed that all had defects in transferrin glycosylation, which caused the disorder to be properly classified as a CDG2. There are 57 confirmed cases to date, but the overall prevalence of the disease is unknown7. Clinical characteristics of PGM1-CDG are often apparent in infancy and may include facial malformations (cleft palate and/or uvula), episodic low blood sugar (hypoglycemia), problems with the heart (cardiomyopathy) and liver (hepatopathy)8. Slow growth and endocrine problems are also common. Muscular problems, such as exercise intolerance and rapid muscle cell break down (rhabdomyolysis), are very common. Neurological symptoms occur in less than half of PGM1-patients, but may consist of mild to moderate intellectual impairment, movement delay, and seizures7,9,10. Patients with mild PGM1-CDG may go undiagnosed into adulthood. A diagnosis can be determined through transferrin analysis and direct assessment of PGM1 enzymatic activity, but definitive diagnosis is often achieved through genetic testing. Dietary galactose therapy has proven effective at treating PGM1-CDG and is especially important for development in younger patients1,3,11–16.
Synonyms
- CDG-It, CDG-1T
- CDG IT, CDG1T
- Congenital disorder of glycosylation type 1t, Congenital disorder of glycosylation type It
- Phosphoglucomutase 1 deficiency; PGM1 deficiency
- Glycogen storage disease XIV
- GSD XIV; GSD 14
Inheritance
PGM1-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The PGM1 gene encodes a mutase enzyme, phosphoglucomutase 1 (PGM1). Mutases are enzymes that facilitate the movement of part of a molecule from one place to another on the same molecule. PGM1 is located in the cytoplasm where it has a pivotal role in glucose metabolism2,3,13. It also affects the generation of the activated sugars involved in the synthesis of glycan chains that may be attached to various proteins and lipids (Figure 1).
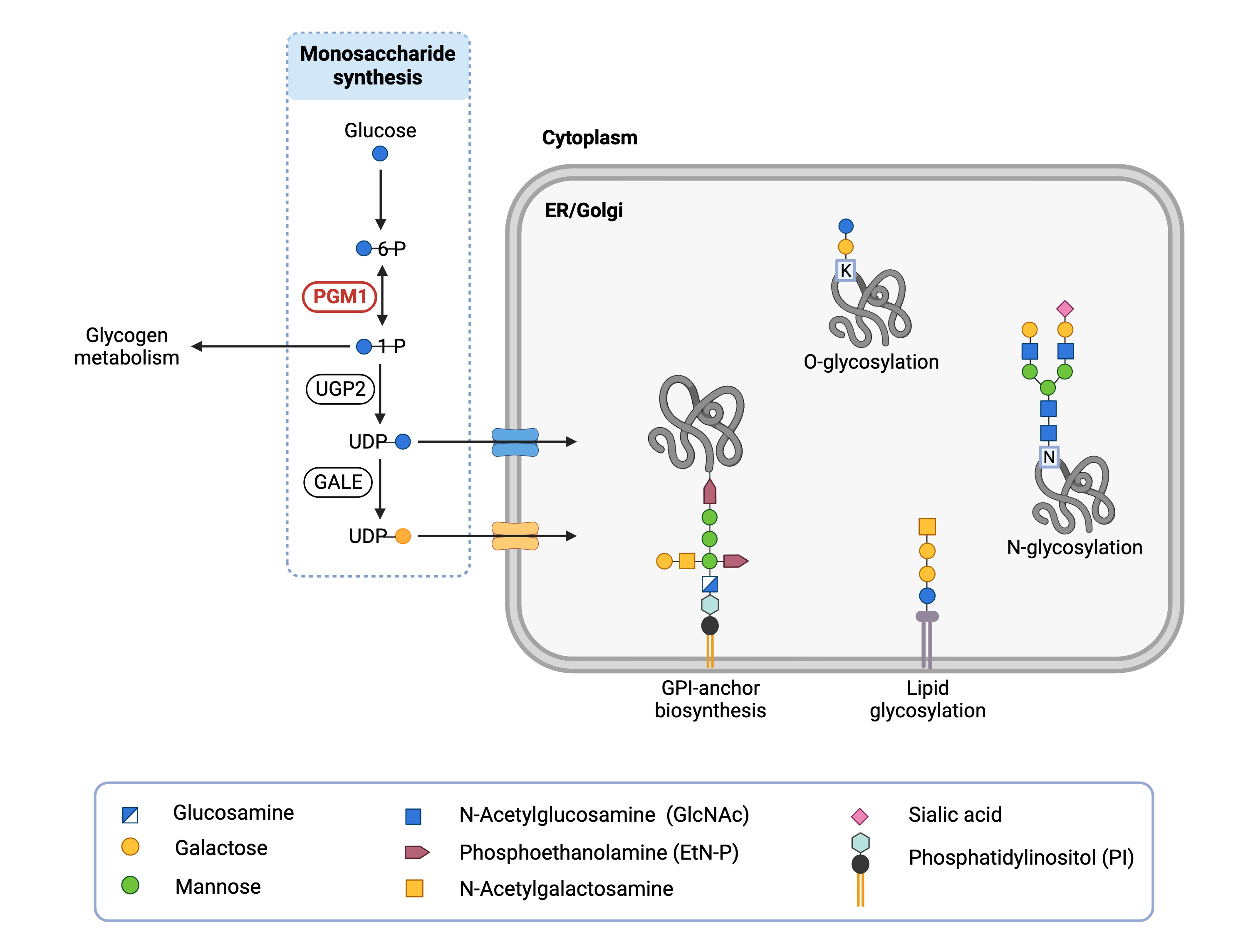
Figure 1. Role of PGM1 in glycosylation.
PGM1 converts glucose-6-phosphate to glucose-1-phosphate, which is used in glycogen metabolism and is needed to make UDP-glucose. UDP-glucose is a precursor for UDP-galactose. PGM1 maintains UDP-glucose and UDP-galactose supply levels, which are used to attach glucose and galactose, respectively, to proteins and lipids in a variety of glycosylation pathways.
Glucose Metabolism
The PGM1 enzyme catalyzes the bidirectional interconversion of glucose-6-phosphate (G6P) and glucose-1-phosphate (G1P). Glucose-1-phosphate, which is formed from the metabolism of sucrose (table sugar), is converted by PGM1 to glucose-6-phosphate. Glucose-6-phosphate is the first intermediate in the conversion of glucose to energy, a process known as glycolysis. In the reverse direction, PGM1 converts glucose-6-phosphate to glucose-1-phosphate, which in turn can be turned into the activated sugar UDP-glucose. In glycogenesis, UDP-glucose is added onto glycogen, a large chain of glucose sugars that is used for energy storage. By facilitating the switch between glucose-1-phosphate and glucose-6-phosphate, PGM1 enables both glucose breakdown (glycolysis) and glucose storage (glycogenesis) and maintains this balance (homeostasis) of glucose metabolism depending on the specific energy needs of the body at a given point in time3,17.
Activated Sugar Synthesis
The monosaccharide glucose is an important component of glycans that are attached to various proteins and lipids. Before glucose can be incorporated into glycans, it must be converted to the activated sugar UDP-glucose in the cytoplasm. By maintaining adequate levels of glucose-1-phosphate, PGM1 facilitates the synthesis of UDP-glucose, which is formed by the action of the enzyme UDP-glucose pyrophosphorylase 2 (UGP2) on glucose-1-phosphate. UDP-glucose is an important precursor for two additional activated sugars: dolichol-P-glucose, the glucose donor inside the ER, and UDP-galactose. UDP-glucose 4-epimerase (encoded by GALE) converts UDP-glucose into UDP-galactose, which is used in the attachment of galactose to glycans. Thus, adequate PGM1 activity is important for maintaining sufficient levels of several different activated sugars.
The activated sugars Dol-P-glucose, and UDP-galactose are particularly important for the synthesis of N-linked glycoproteins. The first step of N-glycosylation is the building of a mannose-rich lipid-linked oligosaccharide (LLO) comprised of a 14-sugar oligosaccharide and dolichol pyrophosphate (Dol-PP). The oligosaccharide component, also referred to as the oligosaccharide precursor, is subsequently transferred en bloc to an asparagine residue of a protein inside the ER. The synthesis of the LLO occurs through a stepwise process, where sugar subunits are added one-at-a-time. The final three sugars that are added prior to its transfer to the protein are glucose residues obtained from Dol-P-glucose. After the oligosaccharide has been transferred to the glycoprotein, the N-glycan undergoes additional modifications in the ER and Golgi, which may include the addition of galactose residues using UDP-galactose.
Disease Mechanism
Mutations in the PGM1 gene lead to the production of an abnormal enzyme with severely reduced activity. PGM1 deficiency prevents the conversion of glycogen, normally stored in the liver and muscle cells, into glucose-6-phosphate which is used to provide energy for cells via glycolysis3,8. This imbalance in glucose metabolism results low blood sugar, which can lead to reduced growth and can be damaging to the brain10. In PGM1 deficiency, muscles cannot rely on glycogen for energy, making it harder to sustain exercise; this can be particularly problematic for the muscles of the heart6,15,18,19. Under conditions of stress, normal PGM1 also binds to a heart muscle protein (ZASP) that is critical for force transduction (e.g. pumping) in order to supply it with constant energy20. Mutations that cause structural changes to PGM1 may prevent this interaction, and inhibition of this interaction is associated with dilated cardiomyopathy.
Other aspects of PGM1-CDG can be explained by the important role of N-glycans in early development. Inadequate PGM1 activity can lead to reduced levels of glucose-1-phosphate. This can lead to reduced levels of several activated sugars that are important for N-glycosylation including UDP-glucose, Dol-P-glucose, and UDP-galactose.
Mutations
The PGM1 gene is located on Chromosome 1 (1p31.3) and 43 mutations have been reported in PGM1-CDG patients, which may lead to truncated or poorly folded proteins with activity between 0.3% to 20% compared to normal baseline17,20–23. Whereas some mutations may directly impact the catalytic activity of PGM1, other mutations that cause improper folding can reduce the solubility of the protein and lead to the formation of protein aggregates. There is no significant correlation between genotype or residual enzyme activity and phenotype7,24.
Signs & Symptoms
Clinical Presentation
Patients with PGM1-CDG may display a wide range of symptoms with varying severity, but the most frequent symptoms are muscle problems and congenital malformations2,7,8. Previously, it was thought that PGM1-CDG was not associated with issues in the central nervous system (CNS), but more thorough analysis of literature reports of patients revealed that CNS symptoms are present in approximately 40% of cases10. Some common features of PGM1-CDG are obvious from birth and in early childhood, but patients with mild disease may go undiagnosed until adulthood.
- Myopathy – low exercise tolerance, muscle weakness, and breakdown of muscle cells upon exercise (rhabdomyolysis)
- Congenital malformations – cleft palate and/or uvula or Pierre-Robin sequence are very common; other abnormal facial features such as prominent forehead, small face, oddly shaped ears, and increased distance between the eyes may be present. Other reported congenital malformations include missing lumbar vertebra, undescended testes, shortened esophagus, and poorly formed or missing anus
- Neurologic – mild to moderate intellectual and learning disability, delayed speech, and seizures
- Cardiac – Potentially fatal dilated cardiomyopathy, heart failure, cardiac arrest, and electrocardiogram (ECG) abnormalities
- Growth – slow growth resulting in shortened height
Biochemical Abnormalties
Biochemical abnormalities frequently observed in individuals with PGM1-CDG include elevated liver enzymes (transaminases), blood clotting (coagulation) abnormalities, elevated creatine kinase, and periodic low blood sugar (hyperinsulinemic and ketotic hypoglycemia)1,7,25. Exercise may cause increased ammonia in the blood (ammonemia) and myoglobin in the urine (myoglobinuria caused by rhabdomyolysis). Other endocrine abnormalities may include low growth hormone, decreased serum insulin-like growth factor-binding protein 3m, low cortisol, hypogonadotropic hypogonadism, delayed puberty, hypothyroidism as evidenced by elevated thyroid stimulation hormone (TSH), and decreased levels of thyroxin-binding globulin (TBG).
Diagnosis
PGM1-CDG should be suspected in any multisystem disorder, especially in infants that present with cleft palate, liver function abnormalities, hypoglycemia, dilated cardiomyopathy, and delayed growth7,8,25. Adults with mild disease may present with exercise intolerance and rhabdomyolysis6,19,20. Screening in suspected patients typically begins with a blood test to analyze serum transferrin isoforms. Direct assessment of PGM1 enzymatic activity may also be used and is often performed after a transferrin analysis displays a mixed type I/II pattern. However, genetic testing is used to definitively diagnose PGM1-CDG.
Transferrin Analysis
Individuals with PGM1-CDG usually show a mixed type I/II pattern by transferrin isoelectric focusing (TIEF) or mass spectrometry analysis of transferrin. These patterns are observed in CDG that arise due to defects in both LLO assembly in the ER and N-glycan processing in the Golgi. In PGM1-CDG, transferrin often has missing glycan chain and/or truncated glycan chains that lack galactose1,8,13. However, patients may display either a type I pattern or a type II pattern, necessitating the use of genetic testing for definitive diagnosis7.
PGM1 Enzyme Activity
Direct analysis of PGM1 activity can be measured in fibroblasts or leukocytes (white blood cells), with patient levels less than 20% of normal7. Red blood cells cannot be used as these cells lack PGM1.
Biomarkers
Transferrin is both a diagnostic and therapeutic biomarker for PGM1-CDG. Transform glycan isoforms have been shown to normalize over time in patients receiving oral d-galactose supplementation32.
Classification
PGM1-CDG is a disorder multiple glycosylation pathways, and more specifically a disorder of monosaccharide interconversion.
Prognosis
PGM1-CDG prognosis can vary depending on the severity of the disease, and some patients may be undiagnosed until adulthood. Congenital malformation and dilated cardiomyopathy are associated with more severe illness24. Cardiac complications have proven fatal for PGM1-CDG patients over a wide age range (early childhood to adulthood) and patients may require a heart transplant7,8,25,26. The oldest PGM1-patient reported in the literature was 49 years old as of 201927.
Management
Management of PGM1-CDG requires a multidisciplinary team and may include combinations of corrective surgery, physical therapy, occupational therapy, cardiological intervention, and speech therapy. IV glucose and oral sucrose administration during exercise can improve exercise tolerance in a PGM1-CDG patient18,19.
Therapies
Unlike most CDG, PGM1-CDG has an effective treatment in the form of galactose supplementation. Other treatments including gene therapy are currently being explored.
Galactose Supplementation
Oral supplementation of galactose between 0.5 – 3 g per kg bodyweight per day (up to 50 g per day) has been successfully used to treat many of the symptoms of PGM1-CDG7. Marked improvement is seen in exercise intolerance, frequency of hypoglycemia, delayed puberty and hypogonadism, growth, and biochemical abnormalities including liver transaminases and clotting factors1,3,11–15,25. Transferrin glycosylation also normalizes with treatment. Galactose supplementation works by replenishing levels of UDP-galactose and UDP-galactose, which are necessary for normal glycosylation in the ER and the Golgi. The additional supplementation of glucose or uridine alongside galactose have been explored as ways to improve exercise tolerance or increase UDP-galactose synthesis, respectively8,16,18. Although concurrent glucose supplementation during exercise is effective, uridine+galactose supplementation has only proven effective at restoring glycosylation in patient fibroblasts8,16. Due to its overall low toxicity and effectiveness, high-purity D-galactose (CERC-801) is being explored by Cerecor for the treatment of PGM1-CDG, and has received fast track designation by the FDA.
Despite its effectiveness at alleviating multiple symptoms and biochemical abnormalities, galactose supplementation is not a perfect treatment. It requires daily large consumption of galactose sugar, and large doses may not always be tolerated. As well, there is a report of the benefits of galactose consumption may wearing off after 18 months of treatment in a PGM1-CDG patient16. Finally, galactose supplementation is ineffective at treating the cardiac manifestations of PGM1, potentially because it is unable to restore the interaction between PGM1 and ZASP in heart muscle tissue25.
Gene Therapy
The Lai lab at the University of Utah Health is currently developing gene replacement therapy for PGM1-CDG.
Research Models
Several PGM1 research models have been generated including yeast, mouse, and human cell lines.
Yeast (S. cerevisiae)
Yeast contains two phosphoglucomutase (PGM1 and PGM2) enzymes that are orthologous to the human enzymes, but PGM2 is the predominant isoform in yeast whereas PGM1 is the dominant isoform in humans28,29.
Pgm2-null mutant
Pgm2 null yeast were viable but demonstrated an increase in glucose-1-phosphate, increased calcium uptake, and somewhat reduced growth when grown on galactose media compared to wildtype yeast29.
Pgm1 pgm2 double null mutant
A Pgm1 Pgm2 double null mutant is viable but cannot use galactose as the sole carbon source. They also accumulate less glycogen and trehalose (a sugar consisting of two glucose subunits) compared to wildtype yeast30.
Mouse (M. musculus)
Whole body constitutive knockout
Murine Pgm2 is orthologous to human PGM1. CRISPR-Cas9 was used to create heterozygous Pgm2+/- mice, which were mated to produce homozygous offspring31. Pgm2 null embryos die before E9.5. Oral galactose supplementation to dams did not rescue the null embryos.
Cardiac specific conditional knockout and gene therapy models
A conditional knockout model that only affects cardiac tissue is being developed in Dr. Kent Lai’s lab at University of Utah Medicine. His lab is also exploring gene therapies for PGM1-CDG in mouse models.
Embryonic stem cell lines
Several embryonic stem (ES) cell lines have been created with mutations in the Pgm2 gene.
Human Cell Lines
Patient-derived fibroblasts
Galactose supplementation in patient derived fibroblasts results in substantially improved protein glycosylation, which is further enhanced by the addition of uridine8.
HeLa cells
A HeLa cell line was used to generate a mammalian two-hybrid model for the co-expression of PGM1 and LDB3; LDB3 encodes an alternatively spliced ZASP protein that is associated with dilated cardiomyopathy20. The cell line was used to study the interaction between PGM1 and ZASP.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including PGM1-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Unknown Status
Study of ORL-1G (D-galactose) in Patients With Glycogen Storage Disease Type 14
Orpha Labs was recruiting patients with PGM1-CDG, previously known as Glycogen Storage Disease Type 14, to study the effect of galactose supplementation on liver function and serum transferrin glycosylation patterns. The status of this trial is unknown.
Fat and Sugar Metabolism During Exercise in Patients with Metabolic Myopathy
Rigshospitalet in Denmark was conducting a study to analyze the metabolic abnormalities that occur during exercise in 21 different metabolic myopathies, including PGM1-CDG. The status of this trial is unknown.
Publications
PGM1-CDG Scientific Articles on PubMed
International consensus guidelines for PGM1-CDG: Diagnosis, follow-up, and management
Additional Resources
PGM1-CDG on FCDGC
IEMbase
OMIM
OrphaNet
GARD
Genetic Testing Registry
ClinVar
NIH
GeneCards
UniProt
References
- Conte, F. et al. Phosphoglucomutase-1 deficiency: Early presentation, metabolic management and detection in neonatal blood spots. Mol. Genet. Metab. 131, 135–146 (2020).
- Pérez, B. et al. A novel congenital disorder of glycosylation type without central nervous system involvement caused by mutations in the phosphoglucomutase 1 gene. J. Inherit. Metab. Dis. 36, 535–542 (2013).
- Radenkovic, S. et al. The Metabolic Map into the Pathomechanism and Treatment of PGM1-CDG. Am. J. Hum. Genet. 104, 835 (2019).
- Thomson, W. H. S., Maclaurin, J. C. & Prineas, J. W. Skeletal muscle glycogenosis: an investigation of two dissimilar cases. J. Neurol. Neuirosurg. Psychiat 26, 68 (1963).
- Sugie, H. et al. Infantile muscle glycogen storage disease: Phosphoglucomutase deficiency with decreased muscle and serum carnitine levels. Neurology 38, 602–605 (1988).
- Stojkovic, T. et al. Muscle Glycogenosis Due to Phosphoglucomutase 1 Deficiency. N. Engl. J. Med. 361, 425–427 (2009).
- Altassan, R. et al. International consensus guidelines for phosphoglucomutase 1 deficiency (PGM1-CDG): Diagnosis, follow-up, and management. J. Inherit. Metab. Dis. 44, 148–163 (2021).
- Tegtmeyer, L. C. et al. Multiple Phenotypes in Phosphoglucomutase 1 Deficiency. N. Engl. J. Med. 370, 533 (2014).
- Ondruskova, N. et al. Glycogen storage disease-like phenotype with central nervous system involvement in a PGM1-CDG patient. Neuro Endocrinol. Lett. 35, 137–141 (2014).
- Radenkovic, S., Witters, P. & Morava, E. Central nervous involvement is common in PGM1-CDG. Mol. Genet. Metab. 125, 200–204 (2018).
- Wong, S. Y. W. et al. Oral D-galactose supplementation in PGM1-CDG. Genet. Med. 19, 1226 (2017).
- Perales-Clemente, E. et al. A new D-galactose treatment monitoring index for PGM1-CDG. J. Inherit. Metab. Dis. 44, 1263–1271 (2021).
- Morava, E. Galactose supplementation in phosphoglucomutase-1 deficiency; review and outlook for a novel treatable CDG. Mol. Genet. Metab. 112, 275 (2014).
- Donoghue, S. E. et al. Galactose treatment of a PGM1 patient presenting with restrictive cardiomyopathy. JIMD Rep. 57, 29–37 (2021).
- Voermans, N. C. et al. PGM1 deficiency: Substrate use during exercise and effect of treatment with galactose. Neuromuscul. Disord. 27, 370–376 (2017).
- Nolting, K. et al. Limitations of galactose therapy in phosphoglucomutase 1 deficiency. Mol. Genet. Metab. Reports 13, 33 (2017).
- Stiers, K. M., Kain, B. N., Graham, A. C. & Beamer, L. J. Induced Structural Disorder as a Molecular Mechanism for Enzyme Dysfunction in Phosphoglucomutase 1 Deficiency. J. Mol. Biol. 428, 1493–1505 (2016).
- Preisler, N. et al. Fat and carbohydrate metabolism during exercise in phosphoglucomutase type 1 deficiency. J. Clin. Endocrinol. Metab. 98, (2013).
- Preisler, N. et al. Impaired glycogen breakdown and synthesis in phosphoglucomutase 1 deficiency. Mol. Genet. Metab. 122, 117–121 (2017).
- Arimura, T. et al. Impaired binding of ZASP/Cypher with phosphoglucomutase 1 is associated with dilated cardiomyopathy. Cardiovasc. Res. 83, 80–88 (2009).
- Beamer, L. J. Mutations in hereditary phosphoglucomutase 1 deficiency map to key regions of enzyme structure and function. J. Inherit. Metab. Dis. 38, 243–256 (2015).
- Stiers, K. M., Graham, A. C., Kain, B. N. & Beamer, L. J. Asp263 missense variants perturb the active site of human phosphoglucomutase 1. FEBS J. 284, 937–947 (2017).
- Lee, Y., Stiers, K. M., Kain, B. N. & Beamer, L. J. Compromised catalysis and potential folding defects in in vitro studies of missense mutants associated with hereditary phosphoglucomutase 1 deficiency. J. Biol. Chem. 289, 32010–32019 (2014).
- Wong, S. Y. W. et al. Defining the Phenotype and Assessing Severity in Phosphoglucomutase-1 Deficiency. J. Pediatr. 175, 130-136.e8 (2016).
- Schrapers, E. et al. News on Clinical Details and Treatment in PGM1-CDG. JIMD Rep. 26, 77–84 (2015).
- Fernlund, E. et al. The congenital disorder of glycosylation in PGM1 (PGM1-CDG) can cause severe cardiomyopathy and unexpected sudden cardiac death in childhood. Forensic Sci. Int. Genet. 43, (2019).
- Tian, W. T. et al. Congenital disorder of glycosylation type 1T with a novel truncated homozygous mutation in PGM1 gene and literature review. Neuromuscul. Disord. 29, 282–289 (2019).
- Csutora, P. et al. Inhibition of phosphoglucomutase activity by lithium alters cellular calcium homeostasis and signaling in Saccharomyces cerevisiae. Am. J. Physiol. - Cell Physiol. 289, 58–67 (2005).
- Fu, L., Miseta, A., Hunton, D., Marchase, R. B. & Bedwell, D. M. Loss of the Major Isoform of Phosphoglucomutase Results in Altered Calcium Homeostasis in Saccharomyces cerevisiae *. J. Biol. Chem. 275, 5431–5440 (2000).
- Boles, E., Liebetrau, W., Hofmann, M. & Zimmermann, F. K. A family of hexosephosphate mutases in Saccharomyces cerevisiae. Eur. J. Biochem. 220, 83–96 (1994).
- Balakrishnan, B. et al. A novel phosphoglucomutase-deficient mouse model reveals aberrant glycosylation and early embryonic lethality. J Inherit Metab Dis 42, 998–1007 (2019).
- Perales-Clemente, E. et al. A new D-galactose treatment monitoring index for PGM1-CDG. J. Inherit. Metab. Dis. 44, 1263–1271 (2021).