
Lay Summary
COG6-CDG, formerly known as CDG-IIL, is a rare inherited condition that affects many parts of the body. To date, 23 cases of COG6-CDG have been reported in the medical literature. COG6-CDG is classified as a disorder of multiple glycosylation pathways. COG6-CDG is caused when an individual has mutations in both copies of their COG6 gene, which provides instructions for making a protein (COG6) needed to help transport glycosylation machinery to the Golgi. Glycosylation machinery, such as enzymes, are delivered to the Golgi in bubble-like structures called vesicles, where they are then involved in glycosylating proteins. COG6 is part of a group of proteins, known as the COG complex, that work together to attach vesicles to the Golgi so that glycosylation machinery can be delivered. Mutations in the COG6 gene cause glycosylation machinery to be delivered to the wrong place in the cell or Golgi which disrupts the two main types of protein glycosylation: N-glycosylation and O-glycosylation. Symptoms of COG6-CDG begin at infancy and are primarily characterized by developmental delay, organ disease, reduced head size, skin abnormalities, and frequent infections. Mutations in the COG6 gene are also associated with Shaheen syndrome, a disorder characterized by intellectual disability, absent sweating, and distinct facial features. Several screening tests are available for COG6-CDG but a definitive diagnosis is achieved through genetic testing. There are currently no approved treatments for COG6-CDG. Treatment is focused on management of specific symptoms and preventing complications.
Overview
Component of Oligomeric Golgi Complex 6 congenital disorder of glycosylation (COG6-CDG) is a rare, autosomal recessive genetic disorder. The first reported case of COG6-CDG was in 20101, and there are 23 confirmed cases to date2. The COG6 gene encodes the COG6 protein, one of the 8 proteins of the conserved oligomeric Golgi (COG) complex. The COG complex is involved in transporting vesicles containing glycosylation enzymes and Golgi proteins to and between compartments of the Golgi; it facilitates the tethering and fusion of vesicles to the Golgi membrane. The COG complex helps ensure that Golgi-resident enzymes and proteins are delivered to the correct compartment of the Golgi. Many proteins require glycosylation to gain full functionality. The first phase of glycosylation involves the attachment of the oligosaccharide to the protein and occurs primarily in the endoplasmic reticulum (ER). These glycoproteins are then shuttled to the Golgi where their glycan chains undergo various modifications performed by different glycosylation enzymes. Mutations in subunits of COG complex result in glycosylation enzymes being delivered to the wrong location, causing defects in N- and O-glycosylation of proteins3,4.
Symptoms of COG6-CDG begin at infancy and the characteristic presentation includes developmental delay, growth problems, facial deformities, and skin problems5. Mutations to the COG6 gene also give rise to Shaheen syndrome, which is characterized by severe intellectual disability, absent sweating, dental problems and thickening of the skin on the hands and feet6. Initial diagnostic tests can be performed to detect abnormal N- and O-glycans in affected individuals7, but a definitive diagnosis of COG6-CDG can only be obtained through genetic testing. No treatment is presently available for treating COG6-CDG.
Synonyms
- CDG syndrome type IIL
- CDG-IIL
- CDG2L
- Congenital disorder of glycosylation type 2l
- Shaheen syndrome
Inheritance
COG6-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene function
The COG6 gene encodes a protein that is subunit 6 of the conserved oligomeric Golgi complex (COG6) — a protein complex made up of a total of 8 subunits. The COG complex plays an important role in ensuring glycosylation enzymes and other Golgi proteins are localized to the appropriate Golgi compartments. The Golgi is made up of multiple compartments, and different steps of a glycosylation pathway may only occur in a particular Golgi compartment. Consequently, proteins required for glycosylation must be delivered to the correct location to ensure proteins are properly glycosylated. Cells use a transportation system, called vesicular trafficking, to move glycosylation proteins between the different Golgi compartments. The COG complex tethers vesicles containing glycosylation proteins to a Golgi compartment, allowing them to be released into the correct Golgi location for glycosylation.
COG6 is one of 8 subunits of the COG complex, all of which are important for the proper functioning of the COG complex and Golgi function3,8. Both N- and O-linked glycosylation of glycoproteins take place within the Golgi.
Vesicular trafficking
Cells use vesicles to transport substances around the cell and move substances between the inside and the outside of a cell — this transportation system is known as vesicular trafficking. Two of the major compartments that vesicles transport between are the endoplasmic reticulum (ER) and the Golgi. The Golgi is a hub for protein and lipid trafficking as well as a site for glycosylation in the cell. The Golgi is made up of three different compartments (cis, medial and trans), which allows different types of glycan modifications to occur in different sections of the Golgi (Figure 1).
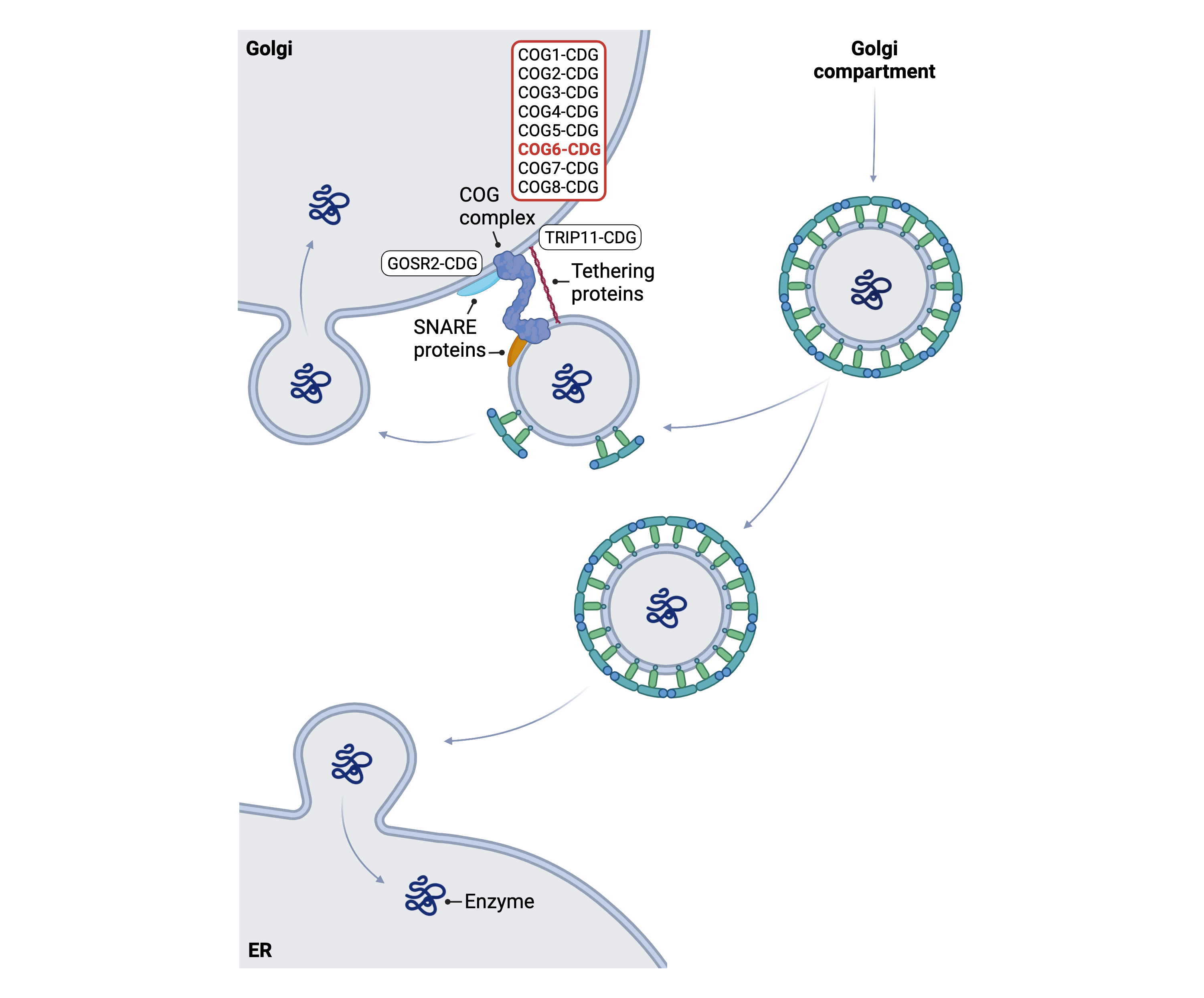
Figure 1. Role of COG6 in glycosylation.
COG6 is a subunit of the conserved oligomeric Golgi (COG) complex, which is needed for lipid and protein glycosylation. The COG complex is needed for vesicles to attach to cell compartment membranes during retrograde vesicular trafficking— a process that allows glycosylation components (such as glycosylation enzymes) to move within and between different cell compartments.
The conserved oligomeric Golgi (COG) complex consists of eight subunits which can be further separated into two lobes: lobe A (containing COG1–4) and lobe B (containing COG5–8). Lobe A mostly associates with the Golgi membrane while lobe B interacts with vesicle membranes9.
The COG complex works with other proteins (such as Rabs, Golgins, and SNARES) to transport glycosylation enzymes and other Golgi-resident proteins (such as sugar transporters) to the correct compartments to ensure correct branching of the glycan structures on a glycoprotein10–12. The COG complex is also involved in the shuttling of vesicles containing cargo from the trans to cis Golgi compartments or from the Golgi to the ER, a process known as retrograde trafficking. The COG complex, along with other tethering proteins, helps tether COPI-coated vesicles carrying glycosylation enzymes to the Golgi membrane. Following tethering, vesicle and Golgi membranes fuse together and the proteins within the vesicle are released into the Golgi10–12.
Disease Mechanism
Mutations in the COG complex subunits disrupt the transport of glycosylation enzymes and cause defective glycan branching of glycoproteins10,11. Mutations causing CDG have been identified in all COG subunits, except for COG3. Loss of functional COG subunits, including COG6, cause the mislocalization of glycosylation enzymes, and disrupts N- and O-linked protein glycosylation in the Golgi10. Variations in one subunit have been shown to cause instability of another subunit, affecting the entire complex10. Although affected individuals with CDG arising from mutations in COG6 and COG7 – which are subunits from the same lobe of the COG complex – can have multiple overlapping clinical features, individuals affected by mutations in COG6 and COG5 (which also both belong to the same lobe) do not have much overlap in clinical features4. Cell studies have suggested that loss of COG6 in the COG complex caused the most severe disruption to Golgi structure13.
The COG complex is involved in the shuttling of other proteins besides those involved in glycosylation and some symptoms of COG6-CDG may be related to other COG6 functions that do not involve glycosylation. Deficiencies in COG also cause fragmented Golgi, abnormal endolysosomes, and general impairment to protein sorting, secretion, and retrograde trafficking; these defects are probably not due to hypoglycosylation but are instead related to other interactions and functions of the COG complex14.
Mutations
The COG6 gene is located on Chromosome 13 (13q14.11) and 13 variants have been reported to date in the COG6 gene. These include 3 splicing variants, 3 nonsense mutations, 5 missense mutations, 1 frameshift mutation resulting from an insertion of base and consequently an early stop codon, and 1 synonymous mutation. With few cases of COG6-CDG documented in the literature, a genotype–phenotype correlation is not yet clear. The splice variant (c.1167–24A>G) has been documented in 1 patient with Shaheen syndrome which is characterized by a milder clinical presentation, whereas nonsense mutations appear to give rise to the most severe clinical presentation in patients4,5.
Signs & Symptoms
Clinical Presentation
Individuals with COG6-CDG typically develop signs and symptoms during infancy. COG6-CDG is primarily characterized by developmental disability, facial structure abnormalities, skin problems, and reduced growth. The characteristic clinical presentations of COG6-CDG include1,2,6,14–19:
- Developmental – failure to thrive, reduced brain volume (cerebral atrophy), inability to control muscles, limbs, and head (psychomotor delay), and reduced or delayed development of mental capacities.
- Dysmorphic features – small head (microcephaly), upturned or tubular nose, misshaped skull, and large mouth
- Growth problems – reduced height and weight and reduced muscle mass (hypotonia)
- Dermatological – thickened skin (hyperkeratosis), dry skin, orange peel skin, excessive sweating (hypohidrosis), red rash, and reduced body hair and ability to sweat
Other observed symptoms of COG6-CDG can include liver and heart defects, sex organ developmental defects, recurrent infections, gastrointestinal defects, structural deformities within fingers, inverted nipples, disorders of sex development, and abnormal reflexes.
Some patients may present with Shaheen syndrome which is characterized by intellectual disability, hypohidrosis, abnormal teeth, and acquired microcephaly6.
Biochemical Abnormalities
Biochemical abnormalities observed in some cases of COG6-CDG include elevation of plasma bilirubin levels and lipase levels, low platelet cell count (thrombocytopenia), and increased number of white blood cells (leukocytosis)4.
Classification
COG6-CDG is classified as a disorder of multiple glycosylation pathways and within this group, it is classified as a disorder of vesicle trafficking.
Under the former CDG classification system, COG6-CDG is classified as a Type II CDG, which arises due to defects in the remodelling of N-glycans once they are attached to proteins.
Diagnosis
Although diagnosis of COG6-CDG may be suspected based on presentation of symptoms and a detailed patient history, direct molecular genetic testing is the only definitive diagnostic test. First-line screening tests in suspected patients are serum transferrin analysis and apolipoprotein C-III analysis, as COG6-CDG can cause defects within both N-linked glycosylation and O- linked glycosylation pathways. Isoelectric focusing analysis of the serum glycoprotein alpha-1 may also be performed. However, genetic testing via next-generation sequencing is required for a definitive diagnosis of COG6-CDG.
Transferrin Analysis
Individuals with COG6-CDG show a type II pattern by transferrin isoelectric focusing (TIEF)4. Type 2 patterns are observed in CDG that arise due to defects after glycan transfer in the ER or during Golgi glycosylation, where remodelling of N-linked glycans occurs.
Apolipoprotein C-III Analysis
COG6-CDG patients can also show an abnormal apolipoprotein C-III isoform pattern through IEF analysis, displaying an abnormal pattern associated with O-linked glycosylation defects1,19.
Alpha-1 Antitrypsin Analysis
COG6-CDG patients can also show abnormal glycosylation patterns by isoelectric focusing analysis of alpha-1 antitrypsin, an abundant serum glycoprotein that exists in different isoforms1.
Biomarkers
A biomarker that is specific to COG6-CDG has not been identified.
Prognosis
Prognosis of COG6-CDG may vary depending on the severity of an individual’s symptoms and the type of mutation they harbor. COG6-CDG can result in death in the first years of life. Of the 21 reported patients with COG6-CDG, seven have died within the first 15 months of their lives4. As of 2021, the oldest living patient with COG6-CDG is 21 years old5.
Management
Management of COG6-CDG focuses on symptoms and avoiding complications. Management of symptoms may include combinations of physical and occupational therapy.
Therapies
There are currently no treatment options available for COG6-CDG.
Research Models
Several research models have been generated to COG6-CDG, including yeast and mouse models and a human cell line.
Yeast (S. cerevisiae)
The yeast COG complex has been studied extensively to delineate the mechanisms of vesicular trafficking. However, yeast missing the COG6 protein grow just as well as their wild-type counterparts, therefore it is unclear whether yeast is an appropriate model organism for studying COG6-CDG8.
Mouse (M. musculus)
COG6 conditional-ready floxed knockout embryonic stem cell line
Embryonic stem (ES) cell line Cog6tm1a(EUCOMM)Wtsi is a targeted knockout/null mutation of Cog6. A “conditional-ready” allele can be created by flp recombinase expression in mice carrying this allele, and cre expression results in a knockout mouse. If cre is expressed without flp expression, a reporter knockout mouse can be generated (MGI). Mice generated with this knockout live to early adulthood, and phenotypically the mutation significantly affects skeletal development, immune and hematopoietic systems, metabolism, skin pigmentation, and vision (IMPC).
Human Cell Lines
HEK293T cells
HEK293T cells with COG subunit knockouts have helped determine the contribution of each subunit to the function and stability of the complex13. Blackburn J, et al. have created a complete set of HEK293T cells with individual COG subunits knocked out, which can be used to characterise glycosylation and trafficking defects. So far, all COG subunit knockouts have been shown to have abnormal Galanthus nivalus lectin (GNL) binding to plasma membrane glycoconjugates, abnormal binding and trafficking of toxins, cis, medial, and trans-Golgi glycosylation defects, abnormal Golgi morphology, and significant fragmentation of the trans Golgi network13. Knockouts of lobe A and COG6 appear to have the most disrupted Golgi structure, suggesting some interactions may be more important for maintaining normal Golgi stacking13.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including COG6-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Publications
COG6-CDG Scientific Articles on PubMed
Additional Resources
OMIM
IEMbase
Orphanet
GARD
NORD
MedlinePlus
Genetic Testing Registry
ClinVar
GeneCards
Uniprot
References
- Lübbehusen, J. et al. Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation. Human Molecular Genetics 19, (2010).
- Komlosi, K. et al. Neonatal presentation of COG6‐CDG with prominent skin phenotype. JIMD Reports 55, 51 (2020).
- Smith, R. D. & Lupashin, V. V. Role of the conserved oligomeric Golgi (COG) complex in protein glycosylation. Carbohydrate Research vol. 343 (2008).
- Rymen, D. et al. Key features and clinical variability of COG6-CDG. Molecular Genetics and Metabolism 116, 163–170 (2015).
- Zhao, P. et al. Genetic analysis and prenatal diagnosis in a Chinese with growth retardation, abnormal liver function, and microcephaly. Molecular Genetics and Genomic Medicine 9, (2021).
- Shaheen, R. et al. A novel syndrome of hypohidrosis and intellectual disability is linked to COG6 deficiency. Journal of Medical Genetics 50, 431–436 (2013).
- Haijes, H. et al. Expanding the clinical phenotype of COG6 deficiency. Journal of Medical Genetics 51, (2014).
- Ishii, M., Lupashin, V. V. & Nakano, A. Detailed analysis of the interaction of yeast COG complex1. Cell Structure and Function 43, (2018).
- D’Souza, Z., Taher, F. S. & Lupashin, V. v. Golgi inCOGnito: From vesicle tethering to human disease. Biochimica et Biophysica Acta - General Subjects 1864, (2020).
- Linders, P. T. A., Peters, E., ter Beest, M., Lefeber, D. J. & van den Bogaart, G. Sugary Logistics Gone Wrong: Membrane Trafficking and Congenital Disorders of Glycosylation. International Journal of Molecular Sciences 2020, Vol. 21, Page 4654 21, 4654 (2020).
- Smith, R. D. & Lupashin, V. v. Role of the conserved oligomeric Golgi (COG) complex in protein glycosylation. Carbohydr Res 343, 2024 (2008).
- Fisher, P. & Ungar, D. Bridging the gap between glycosylation and vesicle traffic. Frontiers in Cell and Developmental Biology 4, 15 (2016).
- Blackburn, J. B., Pokrovskaya, I., Fisher, P., Ungar, D. & Lupashin, V. v. COG Complex Complexities: Detailed Characterization of a Complete Set of HEK293T Cells Lacking Individual COG Subunits. Frontiers in Cell and Developmental Biology 4, 23 (2016).
- Zhao, P. et al. Genetic analysis and prenatal diagnosis in a Chinese with growth retardation, abnormal liver function, and microcephaly. Molecular Genetics and Genomic Medicine 9, (2021).
- Alsubhi, S. et al. Congenital disorders of glycosylation: The Saudi experience. American Journal of Medical Genetics, Part A 173, (2017).
- Li, G. et al. Compound heterozygous variants of the COG6 gene in a Chinese patient with deficiency of subunit 6 of the conserved oligomeric Golgi complex (COG6-CDG). European Journal of Medical Genetics 62, (2019).
- Denecke, J. et al. Congenital disorder of glycosylation type Id: Clinical phenotype, molecular analysis, prenatal diagnosis, and glycosylation of fetal proteins. Pediatric Research 58, (2005).
- Huybrechts, S. et al. Deficiency of subunit 6 of the conserved oligomeric golgi complex (COG6-CDG): Second patient, different phenotype. in JIMD Reports vol. 4 (2012).
- Mandel, H. et al. COG6-CDG: Expanding the phenotype with emphasis on glycosylation defects involved in the causation of male disorders of sex development. (2020) doi:10.1111/cge.13816.