
Lay Summary
COG7-CDG, formerly known as CDG-IIe, is a rare inherited condition that affects many parts of the body. To date, 9 cases of COG7-CDG have been reported in the medical literature. COG7-CDG is classified as a disorder of multiple glycosylation pathways. COG7-CDG is caused when an individual has mutations in both copies of their COG7 gene, which provides instructions for making a protein (COG7) needed to help transport glycosylation machinery to the Golgi. Glycosylation machinery, such as enzymes, are delivered to the Golgi in bubble-like structures called vesicles, where they are then involved in glycosylating proteins. COG7 is part of a group of proteins known as the COG complex that work together to attach vesicles to the Golgi so that their glycosylation machinery can be delivered. Mutations in the COG7 gene cause glycosylation machinery to be delivered to the wrong place in the cell or Golgi which disrupts the two main types of protein glycosylation: N-glycosylation and O-glycosylation. Since many proteins require glycosylation to gain full functionality, disruption of N- and O-glycosylation affects many systems in the body. Symptoms of COG7-CDG may begin during pregnancy or infancy and are primarily characterized by severe developmental delay, facial abnormalities, skin abnormalities, and cardiac problems. Several screening tests are available for COG7-CDG, but a definitive diagnosis is achieved through genetic testing. There are currently no approved treatments for COG7-CDG. Treatment is focused on management of specific symptoms and preventing complications.
Overview
Component of Oligomeric Golgi Complex 7 congenital disorder of glycosylation (COG7-CDG) is a rare, autosomal recessive genetic disorder. The first reported case of COG7-CDG was in 2004, and there are 9 confirmed cases to date1–5. The COG7 gene encodes the COG7 protein, one of the 8 proteins of the conserved oligomeric Golgi (COG) complex. The COG complex is involved in transporting vesicles containing glycosylation enzymes and Golgi proteins to and between compartments of the Golgi; it facilitates the tethering and fusion of vesicles to the Golgi membrane6,7. The COG complex helps ensure that Golgi-resident enzymes and proteins are delivered to the correct compartment of the Golgi. Many proteins require glycosylation to gain full functionality. The first phase of glycosylation involves the attachment of the oligosaccharide to the protein and occurs primarily in the endoplasmic reticulum (ER). These glycoproteins are then shuttled to the Golgi where their glycan chains undergo various modifications performed by different glycosylation enzymes. Mutations in subunits of COG complex result in glycosylation enzymes being delivered to the wrong location, causing defects in N- and O-glycosylation of proteins8–10.
Symptoms of COG7-CDG may begin during pregnancy and infancy and the characteristic presentation includes developmental delay, facial structure abnormalities, skin problems, and cardiac problems1–5. Initial diagnostic tests can be performed to detect abnormal N- and O-glycans in affected individuals, but a definitive diagnosis of COG7-CDG can only be obtained through genetic testing. No treatment is presently available for treating COG7-CDG.
Synonyms
- CDG syndrome type IIe
- CDG-IIe
- CDG2E
Inheritance
COG7-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene function
The COG7 gene encodes a protein that is subunit 7 of the conserved oligomeric Golgi complex (COG7) — a protein complex made up of a total of 8 subunits. The COG complex plays an important role in ensuring glycosylation enzymes and other Golgi proteins are localized to the appropriate Golgi compartments6,7. The Golgi is made up of multiple compartments, and different steps of a glycosylation pathway may only occur in a particular Golgi compartment. Consequently, proteins required for glycosylation must be delivered to the correct location to ensure proteins are properly glycosylated. Cells use a transportation system, called vesicular trafficking, to move glycosylation proteins between the different Golgi compartments. The COG complex tethers vesicles containing glycosylation proteins to a Golgi compartment, allowing them to be released into the correct Golgi location for glycosylation 8–10.
COG7 is one of 8 subunits of the COG complex, all of which are important for the proper functioning of the COG complex and Golgi function. Both N- and O-linked glycosylation of glycoproteins take place within the Golgi6,7.
Vesicular trafficking
Cells use vesicles to transport substances around the cell and move substances between the inside and the outside of a cell— this transportation system is known as vesicular trafficking. Two of the major compartments that vesicles transport between are the endoplasmic reticulum (ER) and the Golgi. The Golgi is a hub for protein and lipid trafficking as well as a site for glycosylation in the cell. The Golgi is made up of three different compartments (cis, medial and trans), which allows different types of glycan modifications to occur in different sections of the Golgi6,7.
The conserved oligomeric Golgi (COG) complex consists of eight subunits which can be further separated into two lobes: lobe A (containing COG1–4) and lobe B (containing COG5–8). Lobe A mostly associates with the Golgi membrane while lobe B interacts with vesicle membranes (Figure 1)6,7.
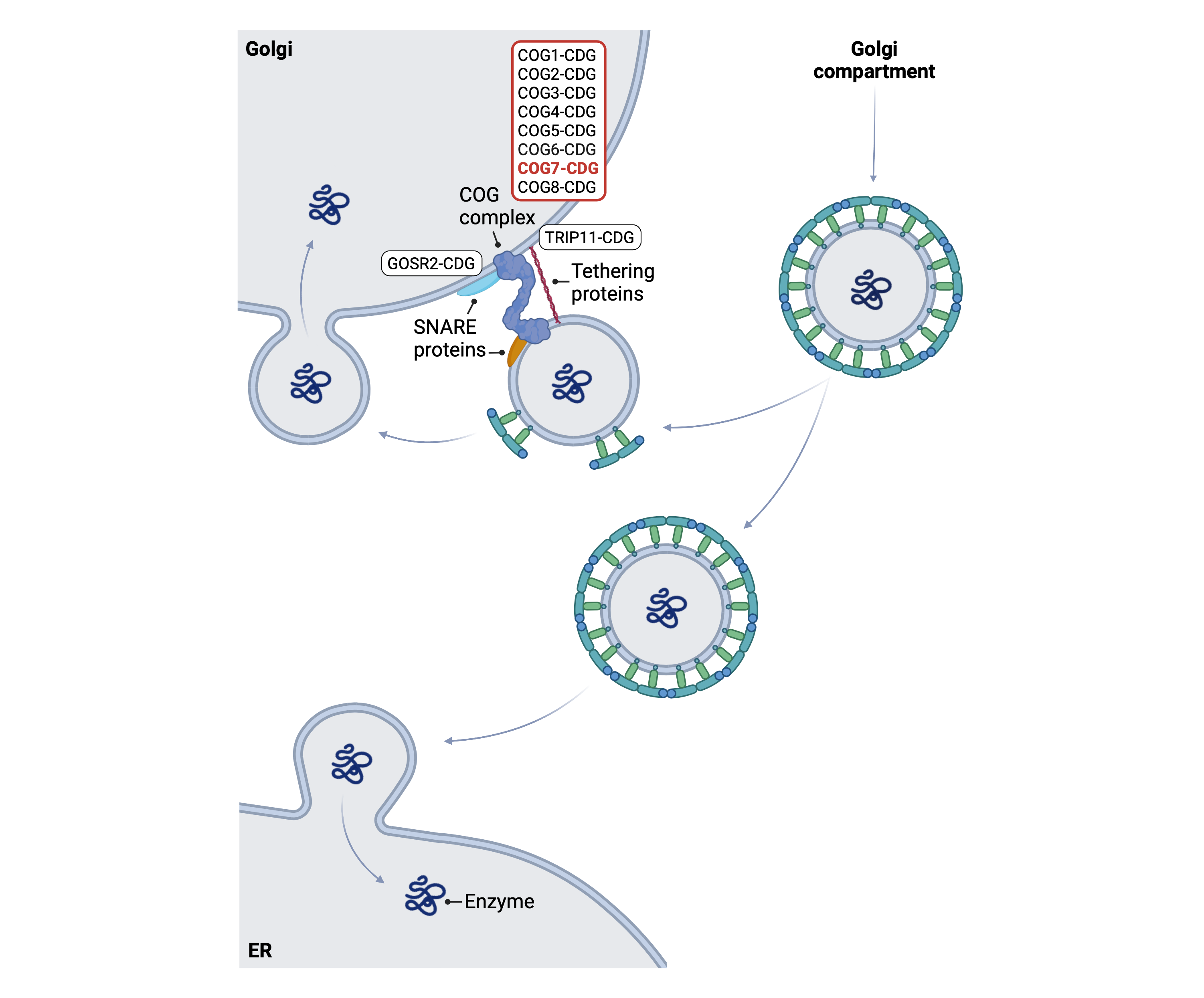
Figure 1. Role of COG7 in glycosylation.
COG7 is a subunit of the COG complex, which is used during retrograde vesicular trafficking. Along with other molecules, such as SNARE and tethering proteins, the COG complex helps tether the vesicle to the Golgi membrane, allowing cargo inside the vesicle to be transported into the Golgi.
The COG complex works with other proteins (such as Rabs, Golgins, and SNARES) to transport glycosylation enzymes and other Golgi-resident proteins (such as sugar transporters) to the correct compartments to ensure correct branching of the glycan structures on a glycoprotein8–10. The COG complex is also involved in the shuttling of vesicles containing cargo the trans to cis Golgi compartments or from the Golgi to the ER, a process known as retrograde trafficking. The COG complex, along with other tethering proteins, helps tether COPI-coated vesicles carrying glycosylation enzymes to the Golgi membrane. Following tethering, vesicle and Golgi membranes fuse together and the proteins within the vesicle are released into the Golgi8–10.
Disease Mechanism
Mutations in the COG complex subunits disrupt the transport of glycosylation enzymes and cause defective glycan branching of glycoproteins. Mutations causing CDG have been identified in all COG subunits, except for COG3. Loss of functional COG subunits, including COG7, cause the mislocalization of glycosylation enzymes, and disrupts N- and O-linked protein glycosylation in the Golgi8. Variations in one subunit have been shown to cause instability of another subunit, affecting the entire complex7.
The COG complex is involved in the shuttling of other proteins besides those involved in glycosylation and some symptoms of COG7-CDG may be related to other COG7 functions that do not involve glycosylation. Deficiencies in COG also cause fragmented Golgi, abnormal endolysosomes, and general impairment to protein sorting, secretion, and retrograde trafficking; these defects are probably not due to hypoglycosylation but are instead related to other interactions and functions of the COG complex11.
Mutations
The COG7 gene is located on Chromosome 16 (16q12.2). Intronic splice site mutation (IVSI+4A>C) and an insertion mutation (p.56-57insAT) have been reported1–5. The patient with the insertion mutation at the protein level was reported to have a longer survival as well as no signs of intrauterine growth retardation or dysmorphic features as clinical features2.
Signs & Symptoms
Clinical Presentation
Individuals with COG7-CDG typically develop signs and symptoms during pregnancy and into infancy. COG7-CDG is primarily characterized by developmental delay, facial structure abnormalities, skin problems, and cardiac problems. The characteristic clinical presentations of COG7-CDG include1–5,12:
- Neurological – profound global developmental delay, seizures, reduced brain volume (cerebral atrophy), and cerebellar atrophy
- Dysmorphic features – small head (microcephaly), low frontal hair line, narrow forehead, micrognathia, retrognathia, smooth philtrum, low set ears, short/wide nose, small mouth, protruding tongue, and short neck
- Growth problems – intrauterine growth retardation, reduced height and weight, reduced muscle mass (hypotonia), and failure to thrive
- Dermatological – loose, wrinkled skin and jaundice
- Cardiac – cardiac insufficiency and cardiac anomalies
Other observed symptoms of COG7-CDG include recurrent infections, liver and kidney defects, skeletal anomalies, gastrointestinal defects, structural deformities within fingers, inverted nipples, and abnormal reflexes.
Biochemical Abnormalities
Biochemical abnormalities observed in some cases of COG7-CDG include abnormal liver enzymes and increased serum creatine kinase12.
Classification
COG7-CDG is classified as a disorder of multiple glycosylation pathways and within this group, it is classified as a disorder of vesicle trafficking.
Under the former CDG classification system, COG7-CDG is classified as a Type II CDG, which arises due to defects in the remodelling of N-glycans once they are attached to proteins.
Diagnosis
Although diagnosis of COG7-CDG may be suspected based on presentation of symptoms and a detailed patient history, direct molecular genetic testing is the only definitive diagnostic test. First-line screening tests in suspected patients are serum transferrin analysis and apolipoprotein C-III analysis, as COG7-CDG can cause defects within both N-linked glycosylation and O- linked glycosylation pathways. However, genetic testing via next-generation sequencing is required for a definitive diagnosis of COG7-CDG.
Transferrin Analysis
Individuals with COG7-CDG show a type II pattern by transferrin isoelectric focusing (TIEF). Type 2 patterns are observed in CDG that arise due to defects after glycan transfer in the ER or during Golgi glycosylation, where remodelling of N-linked glycans occurs2–5.
Apolipoprotein C-III Analysis
COG7-CDG patients can also show an abnormal apolipoprotein C-III isoform pattern through IEF analysis, displaying an abnormal pattern associated with O-linked glycosylation defects2,4,5.
Biomarkers
A biomarker that is specific to COG7-CDG has not been identified.
Prognosis
The prognosis of COG7-CDG may vary depending on the severity of an individual’s symptoms and the type of mutation they harbour but it often results in death in infancy or the first years of life due to severe epilepsy, recurrent infections, cardiac insufficiency and/or respiratory insufficiency1–5.
Management
Management of COG7-CDG focuses on symptoms and avoiding complications. Management of symptoms may include combinations of physical and occupational therapy.
Therapies
There are currently no treatment options available for COG7-CDG.
Research Models
Several research models have been generated for COG7-CDG, including fly, yeast, and mouse models and a human cell line.
Fly (D. melanogaster)
A Drosophila COG7-CDG model has been generated which closely resembles the pathological characteristics of COG7-CDG patients13. Fly’s with COG7 deficiency show pronounced neuromotor defects and altered N-glycome profiles. Decreased lifespan and uncoordinated movement in adult flies along with temperature-sensitive paralysis is reported as well as defects in the development of neuromuscular junctions in larvae. It was also demonstrated that the COG complex regulated Golgi trafficking, associated with Rab1 and Golgi phosphoprotein 3. This Drosophila COG7-CDG model may be used to test therapeutic options for the disease13.
Yeast (S. cerevisiae)
Yeast with mutations to lobe B of the COG complex (COG6, COG7, and COG8) show glycosylation defects as well as defects of internal membrane organization9,14.
Mouse (M. musculus)
COG7 reporter tagged deletion embryonic stem cell line
Embryonic stem (ES) cell line Cog7tm1b(EUCOMM)Hmbu is a reporter tagged deletion mutation of Cog7. Mice generated with this knockout die between E9.5 and early adulthood. This mutation significantly affects heart and eye function in affected mice (IMPC).
Human Cell Lines
HEK293T cells
HEK293T cells with COG subunit knockouts have helped determine the contribution of each subunit to the function and stability of the complex. A complete set of HEK293T cells with individual COG subunits knocked out was created which can be used to characterise the glycosylation and trafficking defects associated with each gene15. So far, all COG subunit knockouts have been shown to have abnormal Galanthus nivalus lectin (GNL) binding to plasma membrane glycoconjugates, abnormal binding and trafficking of toxins, cis, medial, and trans-Golgi glycosylation defects, abnormal Golgi morphology, and significant fragmentation of the trans Golgi network. Knockouts of lobe A and COG6 appear to have the most disrupted Golgi structure, suggesting some interactions may be more important for maintaining normal Golgi stacking15.
COG7-deficient patient fibroblasts
COG7-deficient patient fibroblasts were used to investigate the pathology of Cog7p defects, specifically how the intracellular membrane trafficking pathways are disrupted. Under steady-state conditions, retrograde trafficking, localization, and stability of Golgi proteins were altered in COG7-deficient fibroblasts. COG7-deficient cells also had a delayed response to brefeldin A, a compound that inhibits protein transport, indicating that the COG complex may have a role in Golgi-to-ER transport16.
Co-immunoprecipitation and immunoblotting of Cog7-deficient cells derived from patient fibroblasts were also used to confirm the Cog5-7 subcomplex and the role the COG subunits (1-8) have in the association of the COG complex with the cytoplasmic face of the Golgi17.
Altered endolysosomes and defects in sorting and secretion have been observed in COG7-CDG patient fibroblasts, suggesting that COG deficiencies cause glycosylation-independent defects in Golgi sorting, lysosomal morphology and altered secretion11.
COG7 Knockdown HeLa cells
COG7 knockdown in HeLa cells has been shown to lead to a mislocalization of GlcNAcT1 and GS15, resulting in glycosylation defects along with COG3 knockdown18.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including ALG3-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Publications
COG7-CDG Scientific Articles on PubMed
Additional Resources
OMIM
IEMbase
Orphanet
GARD
NORD
Genetic Testing Registry
ClinVar
GeneCards
References
- Spaapen, L. J. M. et al. Clinical and biochemical presentation of siblings with COG-7 deficiency, a lethal multiple O- and N-glycosylation disorder. J Inherit Metab Dis 28, 707–714 (2005).
- Zeevaert, R. et al. A new mutation in COG7 extends the spectrum of COG subunit deficiencies. European Journal of Medical Genetics 52, 303–305 (2009).
- Ng, B. G. et al. Molecular and clinical characterization of a Moroccan Cog7 deficient patient. Molecular Genetics and Metabolism 91, 201–204 (2007).
- Wu, X. et al. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nature Medicine 10, 518-523 (2004) .
- Morava, E. et al. A common mutation in the COG7 gene with a consistent phenotype including microcephaly, adducted thumbs, growth retardation, VSD and episodes of hyperthermia. European Journal of Human Genetics15, 638–645 (2007).
- D’Souza, Z., Taher, F. S. & Lupashin, V. v. Golgi inCOGnito: From vesicle tethering to human disease. Biochimica et Biophysica Acta - General Subjects 1864, (2020).
- Rabouille, C. et al. Defects in the COG complex and COG-related trafficking regulators affect neuronal Golgi function. 9, 405 (2015).
- Linders, P. T. A., Peters, E., ter Beest, M., Lefeber, D. J. & van den Bogaart, G. Sugary Logistics Gone Wrong: Membrane Trafficking and Congenital Disorders of Glycosylation. International Journal of Molecular Sciences 21, 4654 (2020).
- Smith, R. D. & Lupashin, V. v. Role of the conserved oligomeric Golgi (COG) complex in protein glycosylation. Carbohydr Res 343, 2024 (2008).
- Fisher, P. & Ungar, D. Bridging the gap between glycosylation and vesicle traffic. Frontiers in Cell and Developmental Biology 4, 15 (2016).
- Blackburn, J. B., Kudlyk, T., Pokrovskaya, I. & Lupashin, V. v. More than just sugars: Conserved oligomeric Golgi complex deficiency causes glycosylation-independent cellular defects. Traffic 19, 463–480 (2018).
- OMIM Entry - # 608779 - CONGENITAL DISORDER OF GLYCOSYLATION, TYPE IIe; CDG2E. https://www.omim.org/entry/608779#references.
- Frappaolo, A. et al. COG7 deficiency in Drosophila generates multifaceted developmental, behavioral and protein glycosylation phenotypes. (2017)
- Miller, V. J. & Ungar, D. Re‘COG’nition at the Golgi. Traffic 13, 891–897 (2012).
- Blackburn, J. B., Pokrovskaya, I., Fisher, P., Ungar, D. & Lupashin, V. v. COG Complex Complexities: Detailed Characterization of a Complete Set of HEK293T Cells Lacking Individual COG Subunits. Frontiers in Cell and Developmental Biology 4, 23 (2016).
- Steet, R. & Kornfeld, S. COG-7-deficient Human Fibroblasts Exhibit Altered Recycling of Golgi Proteins □ D. Molecular Biology of the Cell 17, 2312–2321 (2006).
- Oka, T. et al. Genetic analysis of the subunit organization and function of the conserved oligomeric Golgi (COG) complex: Studies of COG5- and COG7-deficient mammalian cells. Journal of Biological Chemistry 280, 32736–32745 (2005).
- Shestakova, A., Zolov, S. & Lupashin, V. COG complex-mediated recycling of golgi glycosyltransferases is essential for normal protein glycosylation. Traffic 7, 191–204 (2006).