
Lay Summary
SLC35C1-CDG, formerly known as CDG-IIc and leukocyte adhesion deficiency type II, is a rare inherited condition that affects many systems in the body. To date, 19 cases have been reported in the medical literature. SLC35C1-CDG is classified as a disorder of multiple glycosylation pathways and is more specifically a disorder of nucleotide sugar transport. SLC35C1-CDG is caused when an individual has mutations in both copies of their SLC35C1 gene which provides instructions for making a protein that transports a high-energy form of the sugar fucose, called a nucleotide sugar to where it is needed in the cell. Mutations in the SLC35C1 gene leads to low levels of fucose in the Golgi Apparatus and insufficient attachment of fucose onto glycosylated proteins (glycoproteins). This particularly impacts some glycoproteins involved in the normal immune response to infection. Symptoms of SLC35C1-CDG begin at infancy or early childhood and are primarily characterized by a compromised immune system, short stature, cognitive impairment, and rare blood type called Bombay blood type. Screening tests are available for SLC35C1-CDG, but a definitive diagnosis is achieved through genetic testing. There are currently no approved treatments for SLC35C1-CDG, however some patients have seen improvements with dietary supplementation of fucose. Recurrent infection appears to improve over time and long-term prognosis is good.
Overview
Solute carrier family 35 member C1 congenital disorder of glycosylation (SLC35C1-CDG) is a rare autosomal recessive genetic disorder. The first cases of SLC35C1-CDG were reported in 19921,2 and there are 19 confirmed cases to date1–17. The SLC35C1 gene encodes the GDP-fucose transporter 1 protein (GFT). GFT is found on the Golgi membrane where it is responsible for transporting the nucleotide sugar GDP-fucose into the Golgi. A sufficient level of GDP-fucose in the Golgi is essential for fucosylation of glycoproteins, and mutations in the SLC35C1 gene result in fucose deficiency in the Golgi and glycoproteins missing fucose residues7. This is particularly important for several fucosylated N-glycoproteins expressed on various blood cell types. For example, the H antigen is a fucosylated glycoprotein that is an intermediate in the synthesis of ABO blood group antigens. As well, sialyl-Lewis X (sLex, CD15s) is a glycoprotein found on the surface of white blood cells (leukocytes) that helps them bind to the walls of blood vessels and pass into areas of infection. Since proper SLC35C1 activity is necessary for normal leukocyte adhesion, SLC35C1-CDG was formerly known as leukocyte adhesion deficiency type II; leukocyte adhesion deficiency disorders are characterized by an inability of leukocytes to leave the vasculature and migrate normally into tissues under conditions of inflammation of infection.
Symptoms of SLC35C1-CDG begin at infancy or early childhood and the characteristic clinical presentation includes a compromised immune system with recurrent infections, short stature, increased white blood cell count, and the rare “Bombay blood type” (hh) caused by a lack of H antigen1,7,18. The compromised immune system is caused impaired leukocyte adhesion and reduced ability to target infections. Due to this, SLC35C1-CDG was formerly known as leukocyte adhesion deficiency type II (LAD II). A diagnosis may be determined through flow cytometry or total N-glycan analysis, as well as testing for H-antigens in the blood, but a definitive diagnosis can only be achieved through molecular genetic testing18. Infection frequency appears to decrease over time and some patients have seen improvements in symptoms when treated with L-fucose (dietary sugar) however there is no FDA-approved treatment at this time18.
Synonyms
- CDG-IIc; CDG 2C
- CDG IIC, CDG2C
- Leukocyte adhesion deficiency type 2
- LAD II
- Rambam-Hasharon Syndrome
Inheritance
SLC35C1-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The solute carrier (SLC) gene superfamily comprises nearly 400 transporter proteins that carry a wide variety of substances across cellular membranes. Within this group, the SLC35 family are transmembrane proteins that function as nucleotide sugar transporters (NSTs)19. Nucleotide sugar transporters transport activated sugars from the nucleus or cytosol into the ER or Golgi, where the sugar component is transferred to glycans of glycoproteins and glycolipids. The SLC35C1 gene encodes the transporter protein, GDP-Fucose transporter 1 (GFT; SLC35C1) which transports GDP-Fucose into the Golgi for fucosylation of glycans19.
Transport of GDP-Fucose
For monosaccharides to be added to proteins and lipids, they must first be turned into more reactive activated sugars, such as nucleotide sugars. Nucleotide sugars are able to donate their sugar component on to other molecules. This process is known as glycosylation and is facilitated by glycosyltransferase enzymes; this process is especially important for the synthesis of glycoproteins in the ER and Golgi. However, nucleotide sugars are synthesized in the cytoplasm and are unable to freely pass through the membranes of the ER and Golgi. Instead, nucleotide sugar transporters embedded in the ER and Golgi membrane transport specific activated sugars into the Golgi and ER, where they can be used in glycosylation.
SLC35C1 is the nucleotide sugar transporter responsible for transporting GDP-fucose into the Golgi. GDP-fucose is made in the cytoplasm and can be formed in several steps from GDP-mannose or by direct activation of fucose sugar20. This direct activation of fucose is called the salvage pathway and is comprised of two main steps – the conversion of fucose into fucose-1-phosphate by fucose kinase and the subsequent conversion of fucose-1-phosphate into GDP-fucose by GDP-fucose pyrophosphorylase.
Once transported inside the Golgi by SLC35C1, GDP-fucose can be used by fucosyltransferase enzymes to transfer fucose onto other molecules (Figure 1). For example, fucose is necessary to form the H-antigen, which is the substructure of the A and B blood group antigens. As well, the sialyl Lewis-X (sLeX) glycan contains a fucose group and is often attached to O-glycans, where is plays a role in cell-to-cell recognition and binding.
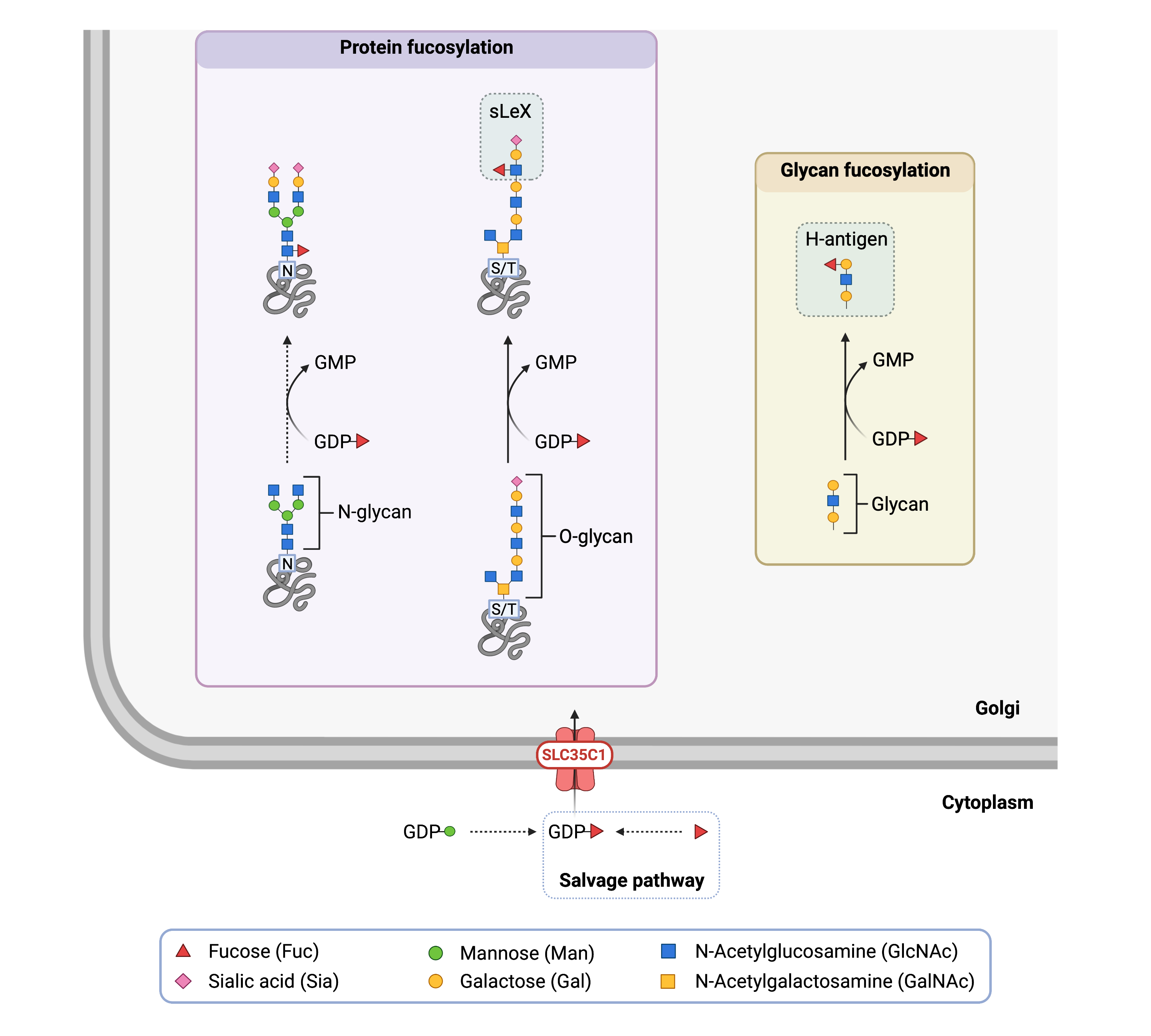
Figure 1. Role of SLC35C1 in glycosylation.
SLC35C1 is nucleotide sugar transporter that transports GDP-fucose into the Golgi from the cytoplasm. Fucosyltransferases can then transfer fucose from GDP-fucose onto proteins, glycans, and lipids.
Disease Mechanism
Mutations in the SLC35C1 gene can result in a protein that has reduced function or is unable to properly localize into the Golgi membrane1. This leads to reduced levels of GDP-fucose in the Golgi and therefore glycans with insufficient fucose residues. For example, lack of properly fucosylated H-antigen leads to the rare Bombay blood type. As well, sialyl Lewis-X is a fucosylated glycan that is important for cell-to-cell recognition, as is required for proper movement of leukocytes through the endothelial barrier of blood vessels into areas of inflammation and infection; Accumulation of leukocytes in these locations results in an influx of neutrophils to help fight the infection. Improperly formed sialyl Lewis-X is responsible for the leukocyte adhesion deficiency phenotype (and former classification) observed in SLC35C1-CDG, and results in reduced ability to fight infections1.
Mutations
The SLC35C1 gene is located on Chromosome 11 (11p11.2) and several mutations in SLC35C1 that give rise to SLC35C1-CDG have been identified to date. The most frequent mutation is homozygous T308R substitution mutation21.
Signs & Symptoms
Clinical Presentation
Individuals with SLC35C1-CDG typically develop signs and symptoms during infancy or childhood. SLC35C1-CDG is primarily characterized by a compromised immune system, neurodevelopment delay, cognitive impairment, and abnormal features1,4,7–18,22.Mild cases of SLC35C1-CDG are less prone to infections.
- Immunological – susceptible to recurrent infections including ear, sinus, and pulmonary infections, infections of the skin or gums, gastroenteritis, and sepsis
- Neurological – developmental delay primarily affecting speech, mild to severe cognitive impairment, low muscle tone (hypotonia), and epilepsy/seizures.
- Behavioural – aggressive behaviour, anxiety, and autism
- Dysmorphic features – short stature, oval face, short nose, and underdeveloped nasal bridge
Other symptoms that have been reported include feeding problems, abnormally small head (microcephaly), astigmatism, kidney swelling due to build up of urine (hydronephrosis), penile malformation (hypospadias), congenital heart defects (atrial septal defect), celiac disease, and delayed teeth development.
Biochemical Abnormalities
Biochemical abnormalities observed in individuals with SLC35C1-CDG include persistent increased levels of neutrophils and decreased levels of B-lymphocytes. The extremely rare blood group called the “Bombay blood type” is also classically associated with SLC35C1-CDG. The Bombay blood type and blood abnormalities may not be present in mild cases1,4,7,10,18.
Classification
SLC35C1-CDG is classified as a disorder of multiple glycosylation pathways, and within this group a disorder of nucleotide sugar transport.
Under the former CDG classification system, SLC35C1-CDG is classified as a Type II CDG, which arise due to which arise due to defects in the processing of N-glycans attached to proteins.
Diagnosis
SLC35C1-CDG should be considered in any multisystemic disorder, especially when a patient presents with marked leukocytosis, severe recurrent infections, the Bombay blood type, short stature, coarse facial features, and cognitive impairment. Diagnostic tests used to diagnose other CDG (e.g., serum transferrin and apolipoprotein CIII analysis) are unremarkable in SLC35C1-CDG. Direct molecular genetic testing is the only way to definitively diagnose the disorder, but total glycan profiling and certain blood biomarkers may aid in diagnosis18.
Total Serum N-glycan Analysis
Analysis of serum total N-glycans by mass spectrometry shows a mild decrease in protein fucosylation10,18.
Flow Cytometry
Patients with SLC35C1-CDG lack CD15/sialyl Lewis-X neutrophil ligands which can be detected using flow cytometry18,23.
Biomarkers
Patients with SLC35C1-CDG lack fucosylated H-antigen and have the Bombay blood type (hh) which may be used for diagnosis18,22.
Prognosis
Life threatening infections are common in early childhood, and approximately 20% of known SLC35C1-CDG patients have died before the age of four9,24. However, infection frequency appears to decrease after the first years of life after which lifespan seems normal. The oldest known patient was 34 at last follow-up7.
Management
Management of SLC35C1-CDG requires a multidisciplinary team and may include combinations of physical therapy, occupational therapy, and speech or vision therapy. Infections typically respond well to antibiotics, and antibiotic prophylaxis can be used in children with very frequent infections24.
Therapies
Oral supplementation of fucose between 0.04–1.5 g per kg bodyweight per day (up to 16 g per day) has been successfully used to treat some patients with both mild and severe cases of SLC35C1-CDG7,9,25,26. Improvements in various blood abnormalities, infection frequency, psychomotor capabilities, and cognitive development (particularly speech) have observed after treatment. Biochemical markers of SLC35C1-CDG, such as H antigen, sialyl Lewis-X, and overall level of fucosylation of serum N-glycoproteins also improved. SLC treated with L-fucose (dietary sugar). Fucose supplementation works by increasing the amount of fucose available for conversion to GDP-fucose through the salvage pathway. This increase in GDP-fucose in the cytoplasm help offset the reduced activity of the SLC35C1 protein seen in most SLC35C1 patients, resulting in higher levels of GDP-fucose entering the Golgi where it can be used in glycosylation. Due to its low overall toxicity and effectiveness, Orpha labs have recently announced recruitment for a Phase III clinical trial evaluating fucose in SLC35C1-CDG. Despite its effectiveness, oral fucose has not resulted in improvements in all patients and has only been tested in a small number of patients. As well, this treatment will not work in cases where SLC35C1 is not incorporated into the Golgi membrane.
Research Models
Several research models have been generated including fly, zebrafish, and mouse models and Chinese hamster ovary cell lines.
Fly (Drosophila melanogaster)
Neurally altered carbohydrate (nac) mutant
The Golgi GDP-fucose transporter (GFR) in flies serves the same functional role as SLC35C1. Some flies demonstrate deficient neural expression of N-glycans with core fucose residues; these flies are known as neurally altered carbohydrate (nac1) mutants27. It was found that this phenotype is caused by loss of activity in GFR that is due to a serine-to-leucine mutation in the protein. The fucosylation defect is resolved when nac1 flies express a wild-type Gfr transgene, indicating that the GFR protein is solely responsible for the fucosylation defect in nac1 mutants. Nac1 flies may be used a model organism to study SLC35C1-CDG since both diseases are caused by inadequate transport of fucose into the Golgi.
Zebrafish (D. rerio)
Slytherin mutant
Zebrafish with a missense point mutation in GDP-mannose 4,6-dehydratase (GMDS), the rate-limiting enzyme in protein fucosylation, are known as slytherin mutants. These mutants display inadequate fucosylation of glycoproteins that results in defects in neuronal differentiation and maintenance, neuromuscular synapse formation, and axon branching; they also display abnormal swimming behavior28. This model was also used to study the roles that hypofucosylation of Notch (a signalling protein) has on the development of the neural phenotype in SLC35C1-CDG. Importantly, the slytherin phenotype was rescued by supplementation with GDP-fucose29. Overall, slytherin mutants are useful for studying abnormal neural development that results from inadequate protein fucosylation, as is caused in SLC35C1-CDG.
Overexpression
To better understand the effect that fucosylation has on embryonic development and various signalling pathways, mouse slc35c1 mRNA was injected into zebrafish embryos, causing over-fucosylation of various glycoproteins and a variety of defects in embryonic development30. It was found that Wnt signalling is moderated by slc35c1 expression via a negative feedback loop. This model may be useful for better understanding the importance of fucose homeostasis in early development.
Chinese hamster Ovary (CHO) cell lines
Slc35c1 knockout (CHO-gmt5)
Derived from MAR-11 mutants, knockout of Slc35c1 in CHO-gmt5 cells results in asialyated and afucosylated proteins due to an absence of a function GDP-fucose and CMP-sialic acid transporter31,32.
Slc35c1 knockout (CHO-gmt3)
Knockout of the Slc35c1 gene by zinc finger nucleotides, TALEN and CRISPR resulted in a lack of functional GDP-fucose transporters and glycoproteins without fucose residues33.
GDP-fucose transporter knockdown (CHO-13D-35D)
Recombinant CHO cells that express human antithrombin III were subjected to a knockdown of the GDP-fucose transporter (GFT) by administering small interfering RNA against CHO GFT or by transfecting the cells with a plasmid expressing the same siRNA. Knockdown of the GFT resulted in a significant decrease in GDP-fucose in the Golgi and an increase in defucosylated antithrombin III34.
Mouse (M. musculus)
Conditional ready knockout mouse or embryonic cell line
Embryonic Stem (ES) cell line Slc35c1tm1a(EUCOMM)Hmgu is a targeted knockout/null mutation of Slc35c1. A “conditional-ready” allele can be created by flp recombinase expression in mice carrying this allele, and cre expression results in a knockout mouse. If cre is expressed without flp expression, a reporter knockout mouse can be generated (MGI).
Slc35c1 knockout mouse ES cells
An in-depth analysis of the glycoproteome was performed in mouse embryonic stem cells and identified that deletion of Slc35c1 resulted in a loss of fucosylated N- and O-glycoproteins and a corresponding increase of the related non-fucosylated glycans. The ablation of fucosylation also conferred resistance to the toxin ricin, as the toxin binds to certain fucosylated glycoproteins35.
Knockout Slc35c1 mouse
Slc35c1-/- mice show a phenotype of severe growth retardation, dilation of lung alveoli, hypocellular lymph nodes and increased postnatal mortality. There was also a significant reduction of fucosylated glycoconjugates in tissues. Fucose treatment only partially normalized the state of glycoproteins36. Additionally, it was found that leukocyte rolling, and neutrophil migration was strongly reduced in Slc35c1-/- mice, similar to clinical phenotype37.
Knockout FX mouse
FX-/- mice with a null mutation in the FX locus, which encodes a GDP-fucose synthesis pathway, present with deficiency in cellular fucosylation and postnatal failure to thrive. FX-/- adult mice phenotypically present similar to LAD-II/CDG-IIc with an absence of leukocyte selectin ligan expression, extreme neutrophilia, and myeloproliferation38.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including ALG3-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Completed
Orpha Labs completed a Phase I/II clinical trial evaluating the safety and efficacy of oral fucose administration in four SLC35C1-CDG patients under the age of 18 (NCT03354533). The primary outcomes were a decrease in frequency of infections and a decrease in neutrophil count.
Publications
SLC35C1-CDG Scientific Articles on PubMed
Additional Resources
SLC35C1-CDG on FCDGC
IEMbase
OMIM
Orphanet
GARD
NORD
Genetic Testing Registry
ClinVar
NIH
Gene Cards
UniProt
References
- Etzioni, A. et al. Recurrent Severe Infections Caused by a Novel Leukocyte Adhesion Deficiency. New England Journal of Medicine. 327, 1789–1792 (2010).
- Frydman, M. et al. Rambam–Hasharon syndrome of psychomotor retardation, short stature, defective neutrophil motility, and bombay phenotype. American Journal of Medical Genetics 44, 297–302 (1992).
- Marquardt, T. et al. Correction of leukocyte adhesion deficiency type II with oral fucose. Blood 94, 3976-3985 (1999).
- Etzioni, A. & Tonetti, M. Leukocyte adhesion deficiency II-from A to almost Z. Immunol Rev 178, 138–147 (2000).
- Etzioni, A. et al. Leukocyte adhesion deficiency (LAD) type II/carbohydrate deficient glycoprotein (CDG) IIc founder effect and genotype/phenotype correlation. Am J Med Genet 110, 131–135 (2002).
- Lübke, T. et al. Complementation cloning identifies CDG-IIc, a new type of congenital disorders of glycosylation, as a GDP-fucose transporter deficiency. Nature Genetics
28, pages73–76 (2001)
- Hüllen, A. et al. Congenital disorders of glycosylation with defective fucosylation. J Inherit Metab Dis 44, 1441–1452 (2021).
- Etzioni, A. & Tonetti, M. Fucose supplementation in leukocyte adhesion deficiency type II. Blood 95, 3641–3 (2000).
- Tahata, S. et al. Defining the mild variant of leukocyte adhesion deficiency type II (SLC35C1-congenital disorder of glycosylation) and response to l-fucose therapy: Insights from two new families and review of the literature. Am J Med Genet A (2022)
- Knapp, K. M. et al. Biallelic variants in SLC35C1 as a cause of isolated short stature with intellectual disability. J Hum Genet 65, 743–750 (2020).
- Cooper, N. et al. Incidental diagnosis of leukocyte adhesion deficiency type II following ABO typing. Clin Immunol221, (2020).
- Yaman, Y. et al. Late diagnosis of leukocyte adhesion deficiency type II and Bombay blood type in a child: a rare case report. Cent Eur J Immunol 44, 206–209 (2019).
- Wolach, B. et al. Leucocyte adhesion deficiency-A multicentre national experience. Eur J Clin Invest 49, (2019).
- Cagdas, D. et al. A novel mutation in leukocyte adhesion deficiency type II/CDGIIc. J Clin Immunol 34, 1009–1014 (2014).
- Dauber, A. et al. Congenital disorder of fucosylation type 2c (LADII) presenting with short stature and developmental delay with minimal adhesion defect. Hum Mol Genet 23, 2880–2887 (2014).
- Helmus, Y. et al. Leukocyte adhesion deficiency II patients with a dual defect of the GDP-fucose transporter. Blood107, 3959–3966 (2006).
- Hidalgo, A. et al. Insights into leukocyte adhesion deficiency type 2 from a novel mutation in the GDP-fucose transporter gene. Blood 101, 1705–1712 (2003).
- SLC35C1 Congenital Disorder of Glycosylation | Rare Diseases Clinical Research Network. https://www.rarediseasesnetwork.org/fcdgc/slc35c1.
- Hadley, B. et al. Nucleotide Sugar Transporter SLC35 Family Structure and Function. Computational and Structural Biotechnology Journal 17, 1123–1134 (2019).
- Becker, D. J. & Lowe, J. B. Fucose: biosynthesis and biological function in mammals. Glycobiology 13, 41R-53R (2003).
- OMIM Entry - * 605881 - SOLUTE CARRIER FAMILY 35, MEMBER C1; SLC35C1. https://www.omim.org/entry/605881.
- Freeze, H. H., Ng, B. G. & Patterson, M. C. Disorders of Glycosylation. Swaiman’s Pediatric Neurology: Principles and Practice: Sixth Edition 317–322 (2017)
- Holland, S. M., Rosenzweig, S. D., Schumacher, R. F. & Notarangelo, L. D. Immunodeficiencies. Infectious Diseases705-722.e2 (2017)
- Etzioni, A. Official reprint from UpToDate Leukocyte-adhesion deficiency. www.uptodate.com (2022).
- Verheijen, J., Tahata, S., Kozicz, T., Witters, P. & Morava, E. Therapeutic approaches in Congenital Disorders of Glycosylation (CDG) involving N-linked glycosylation: an update. Genetics in Medicine 2019 22:2 22, 268–279 (2019).
- Feichtinger, R. G. et al. A spoonful of L-fucose—an efficient therapy for GFUS-CDG, a new glycosylation disorder. EMBO Molecular Medicine 13, e14332 (2021).
- Geisler, C. et al. The Drosophila neurally altered carbohydrate mutant has a defective Golgi GDP-fucose transporter. J Biol Chem 287, 29599–29609 (2012).
- Panzer, J. A. et al. Neuromuscular synaptogenesis in wild-type and mutant zebrafish. Dev Biol 285, 340–357 (2005).
- Song, Y. et al. Neural and synaptic defects in slytherin, a zebrafish model for human congenital disorders of glycosylation. PLoS One 5, (2010).
- Feng, L., Jiang, H., Wu, P. & Marlow, F. L. Negative feedback regulation of Wnt signaling via N-linked fucosylation in zebrafish. Dev Biol 395, 268–286 (2014).
- Haryadi, R., Zhang, P., Chan, K. F. & Song, Z. CHO-gmt5, a novel CHO glycosylation mutant for producing afucosylated and asialylated recombinant antibodies. Bioengineered 4, (2013).
- Zhang, P. et al. Identification of functional elements of the GDP-fucose transporter SLC35C1 using a novel Chinese hamster ovary mutant. Glycobiology 22, 897–911 (2012).
- Chan, K. F. et al. Inactivation of GDP-fucose transporter gene (Slc35c1) in CHO cells by ZFNs, TALENs and CRISPR-Cas9 for production of fucose-free antibodies. Biotechnol J 11, 399–414 (2016).
- Omasa, T. et al. Decrease in antithrombin III fucosylation by expressing GDP-fucose transporter siRNA in Chinese hamster ovary cells. Journal of Bioscience and Bioengineering 106, 168–173 (2008).
- Stadlmann, J. et al. Comparative glycoproteomics of stem cells identifies new players in ricin toxicity. Nature 549, 538–542 (2017).
- Hellbusch, C. C. et al. Golgi GDP-fucose transporter-deficient mice mimic congenital disorder of glycosylation IIc/leukocyte adhesion deficiency II. J Biol Chem 282, 10762–10772 (2007).
- Yakubenia, S. et al. Leukocyte trafficking in a mouse model for leukocyte adhesion deficiency II/congenital disorder of glycosylation IIc. Blood 112, 1472–1481 (2008).
- Smith, P. L. et al. Conditional control of selectin ligand expression and global fucosylation events in mice with a targeted mutation at the FX locus. The Journal of Cell Biology 158, 801 (2002).