
Lay Summary
DPM1-CDG, formerly known as CDG-Ie, is a rare inherited condition that is mainly associated with seizures, developmental delay and small head size at birth. To date, 13 cases of DPM1- CDG have been reported in the literature. DPMI-CDG is classified as a disorder of multiple glycosylation pathways and can be subcategorized as a disorder of dolichol metabolism. DPM1-CDG is caused when an individual has mutations in both copies of the DPM1 gene which provides instructions to for making part of an enzyme that generates the sugar donor molecule dolichol-phosphate-mannose (Dol-P-mannose). Dol-P-mannose is used to donate mannose sugars onto growing sugar chains during protein and lipid glycosylation. Mutations in the DPM1 gene cause some proteins and lipids to have incomplete or absent sugar chains. Common clinical features of individuals affected by DPM1-CDG include developmental delay, seizures, small head size at birth, decreased muscle tone (hypotonia), eye and visual defects, blood clotting defects, and elevated creatine kinase and serum transaminases; these symptoms typically present in infancy. Screening tests are available for DPM1-CDG, but a definitive diagnosis is achieved through genetic testing. Currently, DPM1-CDG does not have any treatments available.
Overview
Dolichol-phosphate-mannose synthase subunit 1 congenital disorder of glycosylation (DPM1-CDG) is a rare autosomal recessive disorder that arises from defects in the DPM1 gene. DPM1 encodes a catalytic subunit of the enzyme: dolichol-phosphate-mannose synthase (DPM synthase)1. DPM synthase transfers mannose from the nucleotide sugar GDP-mannose to dolichol-phosphate (Dol-P), forming Dol-P-mannose2. Dol-P-mannose is a mannose donor used in the construction of the lipid-linked oligosaccharide during N-glycosylation and is also used as a mannose donor in O-glycosylation, C-mannosylation and GPI-anchor protein biosynthesis. Deficiency in DPM1 results in insufficient glycosylation of proteins and GPI-anchor proteins3.
DPM1-CDG was first clinically reported in 20004,5. Altogether, 13 DPM1-CDG cases have been reported in the literature, which have been confirmed by genetic testing1,2,4–8. Onset of symptoms can present from infancy, where common clinical features include developmental delay, seizures, small head size at birth, decreased muscle tone (hypotonia), eye and visual defects, coagulation defects, and elevated creatine kinase and serum transaminases—although symptoms and severity vary between affected individuals1. DPM1-CDG is typically diagnosed through a combination of biochemical (transferrin analysis and alpha-dystroglycan analysis) and genetic testing. As DPM1 affects N-glycosylation, transferrin isoform analysis is usually performed during diagnosis, with cases typically presenting with a type I pattern.1,2. However, genetic testing typically provides a definitive diagnosis.
Synonyms
- CDG syndrome type Ie
- CDG-Ie
- CDG1E
- Carbohydrate deficient glycoprotein syndrome type Ie
- Congenital disorder of glycosylation type 1e
- Congenital disorder of glycosylation type Ie
- Dol-P-mannosyltransferase deficiency
Gene Function
The DPM1 gene encodes dolichol phosphate mannose synthase subunit 1 (DPM1) a subunit of the DPM1 synthase complex. The DPMI complex is located in the ER membrane and is comprised of three subunits, DPM1, DPM2 and DPM1. It is a synthase enzyme which catalyzes the transfer of mannose from the nucleotide sugar GDP-mannose to Dol-P to generate the sugar donor Dol-P-mannose9. Dol-P-mannose is used as a sugar donor in a variety of protein and lipid glycosylation pathways, including O-mannosylation, C-mannosylation, N-glycosylation and GPI-anchor protein biosynthesis (Figure 1)1.
Dol-P-Mannose Synthesis
Dolichol is a lipid that can be found in the membrane of the ER and is important for glycosylation in two ways: it acts a lipid carrier for the 14-sugar oligosaccharide that is transferred onto nascent proteins during N-glycosylation and is a monosaccharide donor for N-glycosylation, O- and C-mannosylation and GPI anchor biosynthesis10.
Dolichol is synthesised through a series of enzymatic reactions in the cytosol and ER membrane. Once dolichol is generated, it must be phosphorylated by the enzyme dolichol kinase (DOLK), generating dolichol-phosphate (Dol-P) before it can be used as a lipid carrier or monosaccharide donor11.
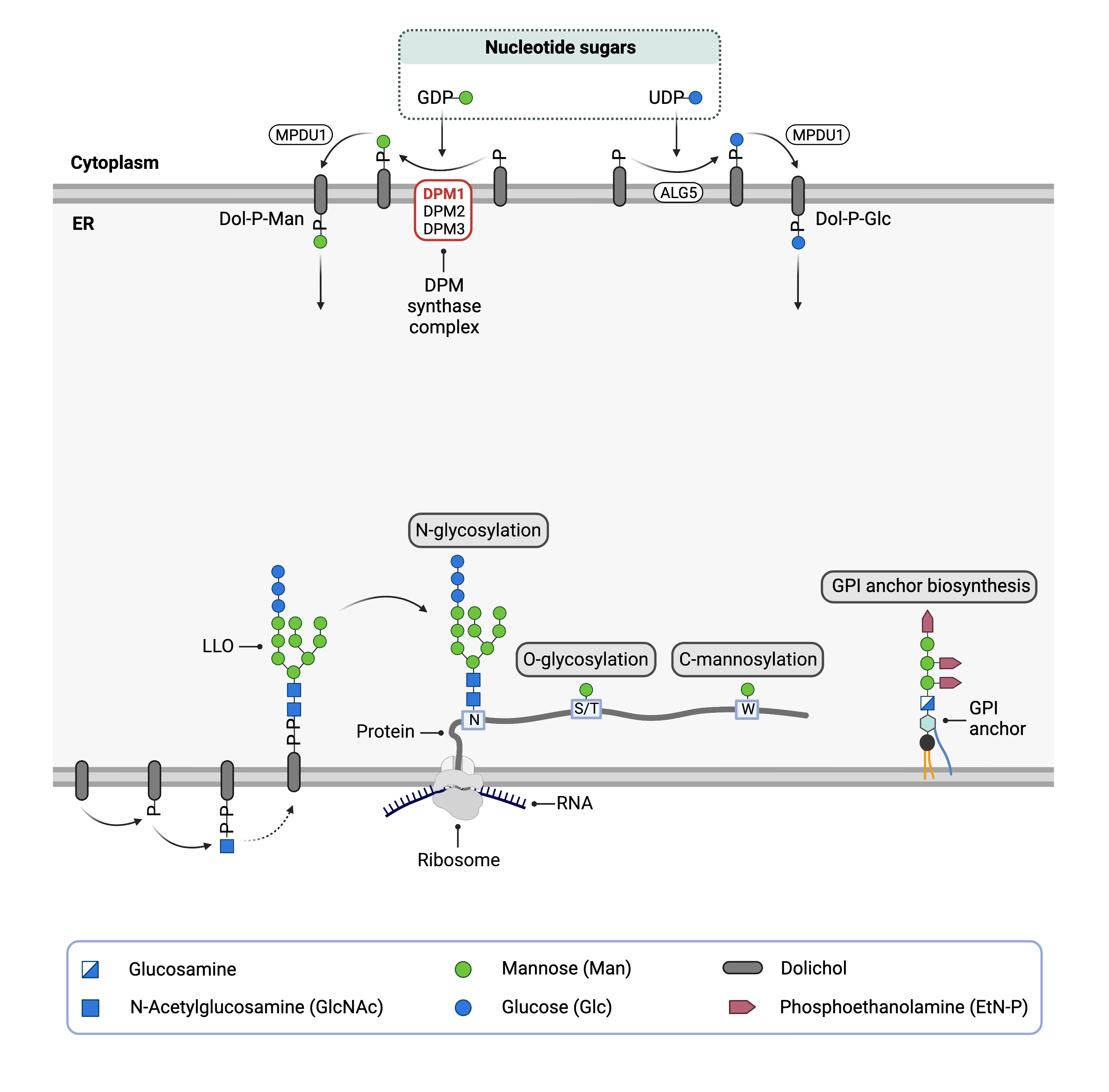
To generate sugar donors Dol-P-mannose and Dol-P-glucose, the monosaccharides must be transferred from nucleotide sugars GDP-mannose and UDP-glucose to Dol-P. The DPM1 synthase complex, comprised of subunits DPM1, DPM2 and DPM3, catalyzes the transfer of mannose from GDP-mannose to Dol-P3. DPM1 is the catalytic domain of the enzyme complex and faces the cytoplasm whereas DPM2 and DPM3 are located in the ER membrane and help anchor and stabilize the complex9.
Dol-P mannose acts a mannose donor for N-, O-, and C-glycosylation and GPI anchor protein biosynthesis. In DPM1-CDG, generation of Dol-P-mannose is impaired and therefore causes defects in multiple glycosylation pathways12.
Disease Mechanism
Defects in DPM1 reduce the ability of DPM synthase to generate Dol-P-Man, which leads to abnormal protein glycans, and potentially GPI-anchored protein glycans2,13. Defective DPM synthase from DPM1 gene mutations leads to DPM synthase having a reduced ability of transferring mannose from GDP-mannose to Dol-P, leading to abnormal glycan structures and levels on proteins2. Pathogenic variants in DPM1 may alter the ability of DPM1 to bind to DPM32.
Mutations
Individuals with DPM1-CDG can be found to have the same (homozygosity) or different mutations in each copy of the DPM1 gene (known as compound heterozygosity). Among the 12 reported cases in the literature, the more common DPM1 pathogenic variants include c.274C>G p.(Arg92Gly) and c.742T>C (p.Ser248Pro). The remaining pathogenic variants from the literature reports include c.331_343del, g.IVS4-5T>A, c.455G>T (p.Gly152Val), exon3–7 deletion, c.1A>C p.(Met1Leu) and c.564-1G>A (p.Leu189Phefs*20)1,2,4–8.
Signs & Symptoms
Clinical Presentation
Affected individuals typically present clinical features from infancy, with common symptoms including developmental delay, seizures, small head size at birth, decreased muscle tone (hypotonia), eye and visual defects and biochemical abnormalities. DPM1-CDG symptoms can include1,2:
- Ophthalmological – uncontrolled eye movements (nystagmus), blindness, optic atrophy, and crossed eyes (strabismus)
- Gastrointestinal – food protein induced enterocolitis syndrome
- Musculoskeletal – hypotonia
- Neurological – seizures, developmental delay, problems with balance and coordination (ataxia), and brain abnormalities
Affected individuals may also have dysmorphic features, such as abnormal nasal bridge, palate and sandal gap1. In addition, affected individuals can display signs of dystroglycanopathy2.
Biochemical Abnormalities
Individuals with DPM1-CDG can have blood clotting (coagulation) defects and elevated liver enzymes (and transaminases) and creatine kinase levels1,2.
Classification
DPM1-CDG is classified as a disorder of multiple glycosylation pathways. More specifically, it is a disorder of dolichol metabolism.
Under the former CDG classification system, DPM1-CDG is classified as a Type 1 CDG4,5, which arise due to defects in the synthesis of N-glycoproteins that occur before the glycan is transferred from the LLO onto the protein.
Diagnosis
DPM1-CDG should be suspected in individuals presenting with developmental delay, seizures, small head size at birth, decreased muscle tone, eye and visual defects 1. Screening in suspected patients typically begins with a blood test to analyze serum transferrin isoforms. As DPM1-CDG can present signs of dystroglycanopathies, muscle biopsies may also be assessed2. However, genetic testing is used to definitively diagnose DPM1-CDG1,2,4–8.
Transferrin Analysis
Individuals with DPM1-CDG show a characteristic type 1 pattern by transferrin isoelectric focusing (TIEF) or mass spectrometry analysis of transferrin14. Type 1 patterns are observed in CDG that arise due to defects in LLO assembly and are characterized by a decrease in tetrasialo-transferrin and an increase in di-sialo and a-sialo transferrin isoforms14.
Muscle Biopsy
Muscle biopsies can be analysed from DPM1-CDG patients using standard histological and immunological staining. In addition, samples can be probed for dystroglycanopathies by staining with anti-dystroglycan antibodies to identify dystroglycan abnormalities2.
Prognosis
There are limited reports of individuals with DPM1-CDG in the medical literature, which makes it difficult to determine the long-term prognosis. From these reports, DPM1-CDG symptoms and disease severity can vary, with a reported case of an affected individual who is 25 years old as of 20181,2,4–8.
Management
There are no therapies currently available for DPM1-CDG, leading treatment to focus on managing symptoms of affected individuals.
Therapies
No therapies currently exist for DPM1-CDG.
Research Models
Several research models have been developed for studying DPM1, including in P. furiosus, yeast, zebrafish, and mice.
Archaea (P. furiosus)
Isolated DPM synthase from P. furiosus
Isolated DPM synthase from P. furiosus has been used in crystallography studies to provide information on the structure and mechanism of DPM synthase, as well as DPM1-CDG15.
Yeast
C. albicans DPM synthase subunit genes expressed in S. cerevisiae
C. albicans DPM synthase genes were expressed in S. cerevisiae. By monitoring gene expression and activity of DPM synthase, experiments showed that the DPM synthase from C. albicans is also made up of multiple subunits16.
T. reesei DPM synthase subunit genes expressed in S. cerevisiae
T. ressei DPM synthase genes were expressed in S. cerevisiae. The model allows the role of each subunit to be probed and analysed17.
Fish (D. rerio)
Depleted DPM1, DPM2 and DPM2 using antisense morpholino oligonucleotides
Antisense morpholino oligonucleotides that depleted DPM synthase subunits were used to model CDG in Zebrafish, showing that DPM synthase provides muscle stability57.
Mouse (M. musculus)
Viable chimeric founder mice with lethal Dpm1 mutation
Using CRISPR-Cas9, viable chimeric founder mice with lethal Dpm1 mutation were developed. These knockout mice can be used to study gene function18.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including ALG1-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Publications
DPM1-CDG Scientific Articles on PubMed
Additional Resources
DPM1-CDG Infographic
IEMbase
OMIM
OrphaNet
GARD
Genetic Testing Registry
ClinVar
NIH
GeneCards
UniProt
References
- Bursle, C. et al. DMP1-CDG (CDG1e) with significant gastrointestinal manifestations; phenotype and genotype expansion. in JIMD Reports vol. 34 (2017).
- Yang, A. C. et al. Congenital disorder of glycosylation due to DPM1 mutations presenting with dystroglycanopathy-type congenital muscular dystrophy. Molecular Genetics and Metabolism 110, 345–351 (2013).
- Maeda, Y. Human dolichol-phosphate-mannose synthase consists of three subunits, DPM1, DPM2 and DPM3. The EMBO Journal 19, 2475–2482 (2000).
- Imbach, T. et al. Deficiency of dolichol-phosphate-mannose synthase-1 causes congenital disorder of glycosylation type Ie. Journal of Clinical Investigation 105, (2000).
- Kim, S. et al. Dolichol phosphate mannose synthase (DPM1) mutations define congenital disorder of glycosylation Ie (CDG-Ie). Journal of Clinical Investigation 105, (2000).
- García-Silva, M. T. et al. Congenital disorder of glycosylation (CDG) type Ie. A new patient. Journal of Inherited Metabolic Disease 27, (2004).
- Medrano, C. et al. Clinical and molecular diagnosis of non-phosphomannomutase 2 N-linked congenital disorders of glycosylation in Spain. Clinical Genetics 95, (2019).
- Dancourt, J. et al. A new intronic mutation in the DPM1 gene is associated with a milder form of CDG Ie in two French siblings. Pediatric Research 59, (2006).
- Lefeber, D. J. et al. Deficiency of Dol-P-Man Synthase Subunit DPM3 Bridges the Congenital Disorders of Glycosylation with the Dystroglycanopathies. American Journal of Human Genetics 85, (2009).
- Cantagrel, V. & Lefeber, D. J. From glycosylation disorders to dolichol biosynthesis defects: a new class of metabolic diseases. Journal of Inherited Metabolic Disease 34, (2011).
- Buczkowska, A., Swiezewska, E. & Lefeber, D. J. Genetic defects in dolichol metabolism. Journal of Inherited Metabolic Disease 38, (2015).
- Ashida, H., Maeda, Y. & Kinoshita, T. DPM1, the catalytic subunit of dolichol-phosphate mannose synthase, is tethered to and stabilized on the endoplasmic reticulum membrane by DPM3. Journal of Biological Chemistry 281, (2006).
- Carmody, L. C. et al. Significantly different clinical phenotypes associated with mutations in synthesis and transamidase+remodeling glycosylphosphatidylinositol (GPI)-anchor biosynthesis genes. Orphanet Journal of Rare Diseases 15, (2020).
- Čechová, A. et al. Consensus guideline for the diagnosis and management of mannose phosphate isomerase‐congenital disorder of glycosylation. Journal of Inherited Metabolic Disease 43, (2020).
- Gandini, R., Reichenbach, T., Tan, T. C. & Divne, C. Structural basis for dolichylphosphate mannose biosynthesis. Nature Communications 8, (2017).
- Juchimiuk, M., Kruszewska, J. & Palamarczyk, G. Dolichol phosphate mannose synthase from the pathogenic yeast Candida albicans is a multimeric enzyme. Biochimica et Biophysica Acta - General Subjects 1850, (2015).
- Zembek, P. et al. Cloning and functional analysis of the dpm2 and dpm3 genes from Trichoderma reesei expressed in a Saccharomyces cerevisiae dpm1Δ mutant strain. Biological Chemistry 392, (2011).
- Wu, Y. et al. Generating viable mice with heritable embryonically lethal mutations using the CRISPR-Cas9 system in two-cell embryos. Nature Communications 10, (2019).