
Lay Summary
NANS-CDG is a rare inherited neurological disorder. To date, 18 cases of NANS-CDG have been reported in the literature. NANS-CDG is classified as a disorder of multiple glycosylation pathways. NANS-CDG is caused when an individual has mutations in both copies of their NANS gene, which provides instructions for making a protein that participates in the producing the sugar sialic acid. Sialic acid can be found at ends of sugar chains (glycans) that are attached to glycosylated proteins or lipids and is essential for normal growth and development of the nervous system, bone, and cartilage. Mutations in the NANS gene cause the enzyme to be inactive and results in insufficient sialic acid levels in the cell. Symptoms of NANS-CDG begin in infancy and are primarily characterized by intellectual developmental delay and abnormal skeletal development. A potential biomarker for NANS-CDG has been identified which may aid diagnosis, but a definitive diagnosis is achieved through genetic testing. There are currently no approved treatments for NANS-CDG. Treatment is focused on the management of specific symptoms and preventing complications.
Overview
N-acetylneuraminic acid synthase congenital disorder of glycosylation (NANS-CDG) is a rare autosomal recessive genetic disorder. The first reported case of NANS-CDG was in 20161, with a total of 18 confirmed cases to date1,2.
N-acetyl-D-neuraminic acid is commonly referred to as sialic acid. Sialic acid is typically found at the end of glycan chains on glycoproteins or glycolipids2. Sialyated (sialic acid-modified) glycoconjugates are abundantly present in the central nervous system, particularly on gangliosides and neural cell adhesion molecules, playing key roles in cell migration, synaptic activity, neural path finding, neurite outgrowth, and regeneration1–4.
Sialic acid is synthesised in the cell through a series of enzymatic reactions. Among these enzymes is NANS (encoded by the NANS gene), which converts N-acetylmannosamine-6-phosphate (ManNAc-6-P) to N-acetylneuraminic acid-9-phosphate (NeuNAc-9-P). A second enzyme (NANP) then converts NeuNAc-9-P into sialic acid, ready for attachment to CMP and allowing sialic acid to be transferred onto proteins and lipids5. Mutations in the NANS gene result in an inactive synthase enzyme, along with reduced levels of sialic acid in the cell and under-sialyated proteins1,2.
Symptoms of NANS-CDG begin at infancy and the characteristic presentation includes intellectual developmental disorder with delayed developmental milestones and abnormal skeletal development (skeletal dysplasia), such as a short stature with short limbs1,2. Elevated ManNAc levels in urine have been observed in NANS-CDG patients and may serve as a diagnostic biomarker2, however, a definitive diagnosis can only be achieved through genetic sequencing. While a clinical trial recently investigated oral supplementation of sialic acid in NANS-CDG patients showed that sialic acid supplements were well tolerated, initial findings did not show clinical benefits to patients5. There are currently no approved treatments for NANS-CDG1,2.
Synonyms
- N-acetylneuraminic acid synthase congenital disorder of glycosylation
- NANS deficiency
- NANSd
- Spondyloepimetaphyseal dysplasia
- Camera-Genevieve type
Inheritance
NANS-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The NANS gene encodes N-acetylneuraminic acid synthase (NANS), an enzyme that catalyzes the synthesis of N-acetylneuraminic acid (NeuNAc; sialic acid) in the cytoplasm. Sialic acid is found on the end of glycan chains on glycoproteins and glycolipids and is abundant in the central nervous system where it is essential for cell migration, synaptic activity, neural path finding, neurite outgrowth, and regeneration. As such, synthesis of sialic acid is essential for early brain development, in addition to having roles in inflammation, neutralization of reactive oxygen species, and neurodegeneration2.
Sialic Acid Synthesis
The synthesis of sialic acid occurs in the cytoplasm, with sialyation of glycoproteins and glycolipids occurring in the Golgi. The enzyme GNE catalyzes the first, rate-limiting step of this process, where the activated sugar UDP-GlcNAc is converted to the sugar ManNAc. Next, GNE adds phosphate to ManNAc, generating ManNAc-6-P. NANS then converts ManNAc-6-P to NeuNAc-9-P, which is converted into sialic acid by the enzyme NANP (Figure 1).
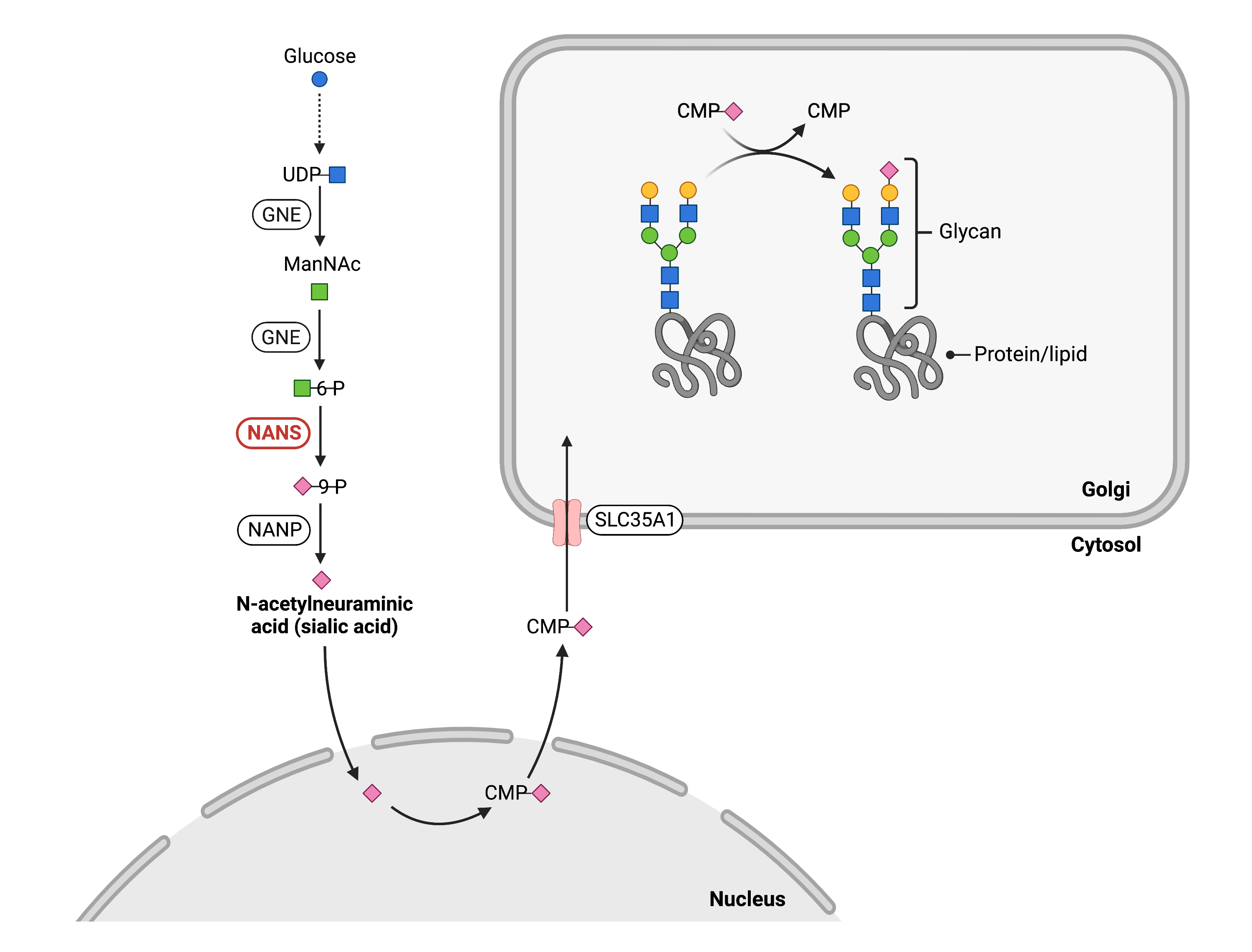
Figure 1. Role of NANS in glycosylation.
NANS is an enzyme that is needed for sialic acid synthesis, which is a monosaccharide that can be used in protein and lipid glycosylation pathways. NANS converts ManNAc-6-phosphate to sialic acid-9-phosphate in the cytosol.
Once sialic acid is synthesized, it is attached to CMP by CMP-sialic acid synthase in the nucleus, generating CMP-sialic acid. CMP-sialic acid is then imported into the Golgi by the CMP-sialic acid transporter SLC35A1. In the Golgi, sialyltransferases facilitate the transfer of sialic acid from CMP-sialic acid onto the terminal ends of N-glycans, O-glycans, glycosphingolipids, or GPI anchor side chains6. Thus, reduced levels of sialic acid that result from deficient NANS activity can affect all major glycosylation pathways4.
Disease Mechanism
Mutations in the NANS gene lead to an inactive synthase enzyme and a reduction of sialic acid in cells1. Sialic acid is essential for normal brain growth and development as it has a role in cell migration, synaptic activity, neural path finding, neurite outgrowth, and regeneration2. The sialic acid residues on gangliosides and glycoproteins in the frontal cortex specifically play an important role in structure and function3,4. Additionally, sialic acid is important for normal skeletal development as several factors in cartilage and bone development are highly sialylated1. Deficient NANS leads to an accumulation of ManNAc and decreased levels of sialic acid in the body, affecting glycans on lipids and proteins. Reducing sialic acid levels also impact heavily sialylated factors of cartilage and bone development and result in the skeletal dysplasia phenotype1.
Mutations
The NANS gene is located on chromosome 9 (9q22.33). Several mutations have been reported, the majority of which are compound heterozygous mutations. Patients with pathogenic variants c.709C>T and c.562T>C are considered to be at the severe end of the phenotypic spectrum, displaying extensive neurological and skeletal anomalies2.
Signs & Symptoms
Clinical Presentation
Individuals with NANS-CDG typically develop signs and symptoms during infancy. NANS-CDG is primarily characterized by severe developmental delay and skeletal dysplasia. Common symptoms of NANS-CDG include1,2:
- Neurological – intellectual developmental disorder with delay in developmental milestones, neurologic impairment, low muscle tone (hypotonia), excess muscle tone (hypertonia), lack of muscle control and coordination (ataxia), and epileptic seizures
- Dysmorphic features – short stature with short limbs, skeletal malformations (skeletal dysplasia), sunken/wide nasal bridge, prominent forehead, and prominent mouth
- Gastrointestinal – feeding difficulties, failure to thrive and severe constipation with abdominal distention
Less common symptoms of NANS-CDG include ophthalmological problems such as strabismus, hearing problems, urinary tract abnormalities, and predilection toward recurrent infections.
Biochemical Abnormalities
Low levels of LDL cholesterol have been detected in some patients, as well as variable levels of alkaline phosphatase (mildly elevated to decreased)2. Accumulation of ManNAc in body fluids has also been reported1.
Classification
NANS-CDG is classified as a disorder of multiple glycosylation pathways and is subcategorized as a disorder of monosaccharide synthesis.
Diagnosis
Individuals with NANS-CDG can present a range of clinical features, with variable disease severity. So far, common reported clinical features of patients with NANS-CDG include intellectual disabilities, neurological defects, skeletal dysplasia, and physical features (such as short limbs and stature and facial dysmorphisms)7. Routine first-line tests, such as transferrin isoform analysis, are not suitable for NANS-CDG diagnosis as individuals can present normal transferrin isoelectric focusing (TIEF) results8,9. Consequently, NANS-CDG diagnosis is achieved through genetic testing7.
Biomarkers
N-Acetylmannosamine (ManNAc)
ManNAc in plasma and urine has been identified as a potential biomarker for NANS-CDG. ManNAc excretion levels have been shown to significantly correlate with phenotypic severity1,2.
Prognosis
Prognosis of NANS-CDG may vary depending on the severity of an individual’s symptoms. With the majority of reported NANS-CDG patients in the medical literature being young, the long-term prognosis is difficult to determine.
Management
Management of symptoms may include combinations of physical therapy, occupational therapy, and palliative measures. Affected patients may require genetic counselling and personalized treatment for different clinical features, including gastrointestinal abnormalities, epilepsy, low platelet counts, and cognitive and physical impairments7.
Therapies
There are currently no approved treatment options available for NANS-CDG. Initial findings from a recent clinical trial (NCT03545568) investigating NANS-CDG treatment with oral sialic acid supplementation showed good tolerability but no clinical benefits to patients1,2,5.
Research Models
Several NANS research models have been generated including zebrafish, mice, and cell lines.
Fish (D. rerio)
Knockdown of nansa in zebrafish embryos resulted in abnormal skeletal development, such as small head size, and pericardial edema. Mutants also showed a complex head phenotype including hypoplastic or absent Meckel’s cartilage. The skeletal phenotype was partially rescued by adding sialic acid exogenously1.
Mouse (M. musculus)
Conditional knockout embryonic stem cell line
Nanstm1a(EUCOMM)Hmgu are mice embryonic stem cell lines with targeted null/knockout mutations of the first allele of Nans (MGI).
Nans exon deletion mouse
Nansem1(IMPC)Tcp mice contain an exon deletion which significantly effects metabolism, vision, growth, and body size, and causes early death. In mice with heterozygous deletions, there is an increased body fat, decreased lean body mass, and cataracts reported. In mice with homozygous deletions, embryonic lethality prior to tooth bud stage, prenatal lethality prior to heart atrial septation, and pre-weaning lethality with complete penetrance is reported (MGI).
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including NANS-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Completed
Sialic Acid Supplementation in N-Acetylneuraminic Acid Synthase (NANS) Deficiency (NCT03545568)
The University of Lausanne and University of Modena and Reggio Emilia conducted an interventional study, investigating the short-term effects of sialic acid supplementation on sialic acid metabolism biomarkers in NANS-CDG patients.
Publications
NANS-CDG Scientific Articles on PubMed
Additional Resources
NANS-CDG Infographic
IEMbase
OMIM
ClinVar
NIH
GeneCards
UniProt
References
- van Karnebeek, C. D. M. et al. NANS-mediated synthesis of sialic acid is required for brain and skeletal development. Nature Genetics 2016 48:7 48, 777–784 (2016).
- den Hollander, B. et al. NANS-CDG: Delineation of the Genetic, Biochemical, and Clinical Spectrum. Frontiers in Neurology 12, (2021).
- Wang, B. Sialic Acid Is an Essential Nutrient for Brain Development and Cognition. Annu Rev Nutr, 177–222 (2009).
- Wang, B. & Brand-Miller, J. The role and potential of sialic acid in human nutrition. European Journal of Clinical Nutrition 57, 1351–1369 (2003).
- Tran, C. et al. The fate of orally administered sialic acid: First insights from patients with N-acetylneuraminic acid synthase deficiency and control subjects. Molecular Genetics and Metabolism Reports 28, (2021).
- Varki, A. & Schauer, R. Sialic Acids. in Essentials of Glycobiology (eds. Varki, A., Cummings, R. D. & Esko, J. D. et al.) 335–349 (Cold Spring Harbor Laboratory Press, 2009).
- den Hollander, B. et al. NANS-CDG: Delineation of the Genetic, Biochemical, and Clinical Spectrum. Frontiers in Neurology 12, (2021).
- van Karnebeek, C. D. M. et al. NANS-mediated synthesis of sialic acid is required for brain and skeletal development. Nature Genetics 48, (2016).
- Freeze, H. H. Perhaps a wee bit of sugar would help. Nature Genetics vol. 48 (2016).