
Lay Summary
PIGA-CDG, also known as PIGA deficiency and Multiple Congenital Anomalies-Hypotonia-Seizures syndrome type 2 (MCAHS2), is a rare inherited condition that affects many parts of the body. It is one of the more common congenital disorders of glycosylation (CDG) with more than 100 cases reported to date in the medical literature. PIGA-CDG is classified as a disorder of GPI anchor biosynthesis. PIGA-CDG is caused when an individual has a mutation in one copy of their PIGA gene (located on the X chromosome) and occurs primarily in males. The PIGA gene provides instructions for making a protein that participates in building GPI anchors which are molecules that “anchor” certain proteins to the cell surface. The PIGA protein is involved in the first step of GPI anchor synthesis where it attaches the simple sugar N-acetylglucosamine (GlcNAc) to the growing GPI anchor. Mutations in the PIGA gene cause defects in GPI-anchored proteins which make them unstable or unable to attach to the surface. Symptoms of PIGA-CDG begin at infancy and are primarily characterized by developmental delay, epilepsy, and low muscle tone. Behavioral abnormalities, skin problems, and gastrointestinal issues may also be present in some individuals with this disorder. Individuals diagnosed with PIGA-CDG often have a lower life expectancy, many of whom pass away at a very young age due to respiratory complications. PIGA-CDG is usually diagnosed through genetic testing, however testing for the presence of GPI-anchored proteins on certain blood cells can also identify PIGA-CDG. With the exception of ketogenic diets which may reduce seizures, there are currently no approved treatments for PIGA-CDG. While there are several therapies in development, currently treatment is focused on the management of specific symptoms and preventing complications.
Overview
Phosphatidylinositol glycan class A congenital disorder of glycosylation (PIGA-CDG) is a rare X-linked recessive genetic disorder. The first reported case of PIGA-CDG was in 2012, and over 100 confirmed cases have been reported to date, making it one of the more commonly diagnosed CDG1–3. PIGA-CDG is commonly referred to as PIGA deficiency or Multiple Congenital Anomalies-Hypotonia-Seizures syndrome type 2 (MCAHS2).
The PIGA gene encodes a subunit of the enzyme complex that catalyzes the first step of glycosylphosphatidylinositol (GPI) anchor protein biosynthesis. GPI anchors are an important mechanism of attachment for proteins to the cell surface, and these proteins are known as GPI-anchored proteins. Mutations in PIGA result in defects in GPI-anchored proteins, which are critical to the development and function of many organs, especially the nervous system2. Somatic mutations in PIGA have been associated with Paroxysmal Nocturnal Hemoglobinuria (PNH), a rare acquired blood disorder4,5.
As PIGA-CDG is an X-linked recessive disorder, symptoms typically only present in males2. Symptoms begin at infancy and the characteristic presentation includes developmental delay, seizures, facial abnormalities, gastrointestinal and breathing problems, as well as decreased muscle tone2,6,7. Although a definitive diagnosis can only be achieved through genetic sequencing, analyzing blood cells for an absence of GPI-anchored proteins can help diagnose disorders of GPI-anchor biosynthesis. No treatment is currently available for PIGA-CDG, although small molecule and gene therapies are in pre-clinical stages8.
Synonyms
- PIGA deficiency
- Multiple Congenital Anomalies-hypotonia-seizures syndrome type 2
- MCAHS2
- Phosphatidylinositol glycan class A congenital disorder of glycosylation
- Ferro-Cerebro-Cutaneous syndrome
- Early infantile epileptic encephalopathy type 20
Inheritance
PIGA-CDG is an X-linked recessive disorder, meaning an affected male inherits a single defective copy of the gene from an asymptomatic (carrier) mother 2.
Gene Function
The PIGA gene encodes the catalytic subunit (PIG-A) of the glycosylphosphatidylinositol (GPI) N-acetylglucosamine transferase complex, which is a glycosyltransferase comprised of 7 proteins. Glycosyltransferases are enzymes responsible for the initiation and elongation of glycan chains.
The PIG-A enzyme is located in the endoplasmic reticulum (ER) and catalyzes the first step of GPI anchor biosynthesis: the transfer of N-acetylglucosamine (GlcNAc) from the nucleotide sugar UDP-GlcNAc to the to the lipid phosphatidylinositol (PI), generating N-acetylglucosaminyl phosphatidylinositol (GlcNAc-PI).
GPI-Anchored Protein Biosynthesis
GPI-anchored protein biosynthesis is one of the major glycosylation pathways that attach glycans to lipid molecules within cells. Many proteins are attached to the cell surface by GPI anchors, which are referred to as GPI-anchored proteins.
The core structure of GPI consists of phosphatidylinositol (PI), glucosamine (GlcN), three mannose sugars (Man3), and phosphoethanolamine (EtN-P) connected to each other in that sequence (Figure 1). GPI-anchored proteins are attached to the GPI by forming a bond between the EtN-P group of the GPI core and the C-terminus of newly synthesized proteins in the ER lumen9.
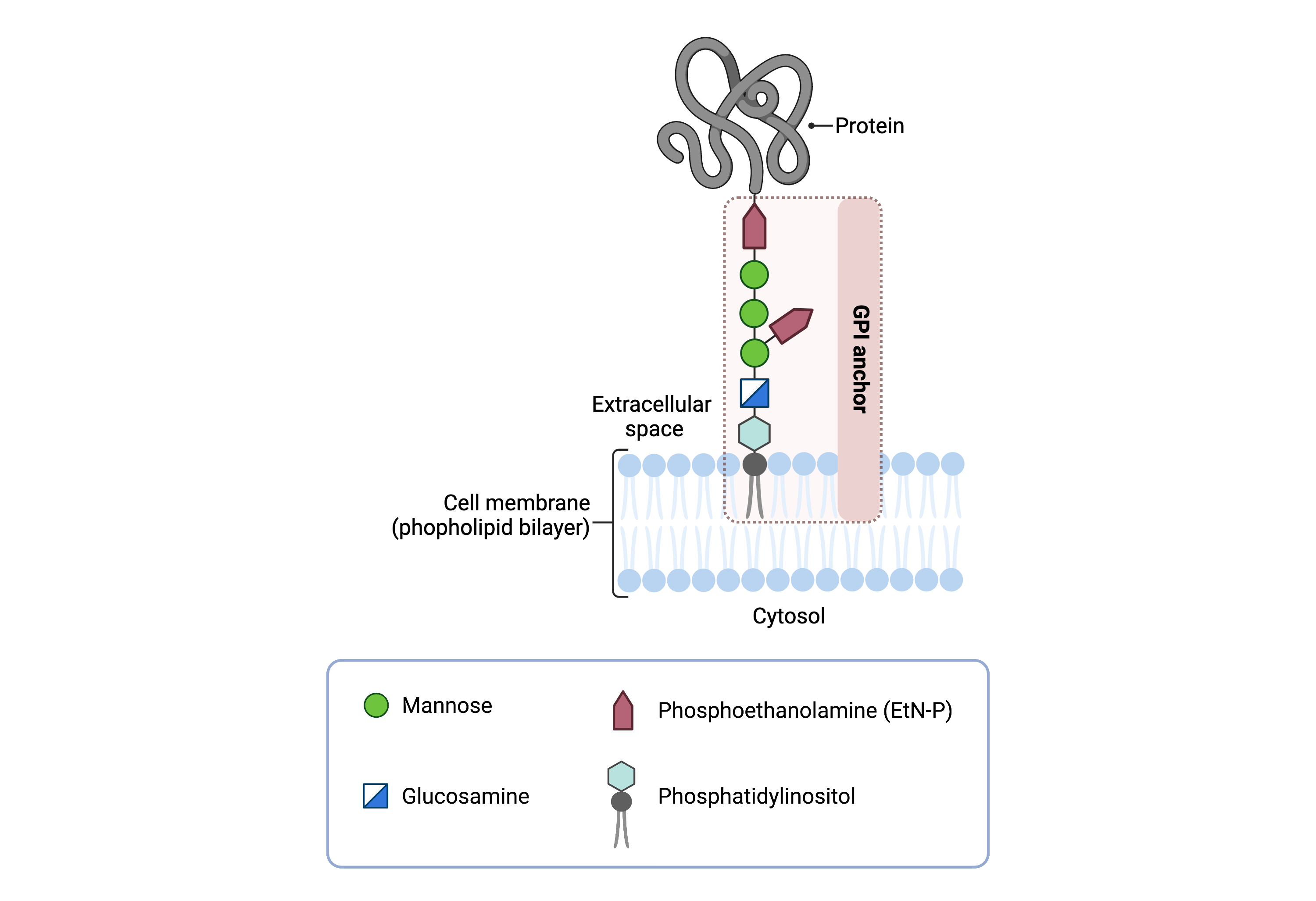
Figure 1. Overview of GPI-anchored protein structure.
The GPI anchor is added to proteins, generating GPI-anchored proteins. The GPI anchor section lodges into the membrane, attaching the protein to the membrane. The GPI anchor involves phosphatidylinositol, glucosamine, mannose and phosphoethanolamine.
The generation of GPI-anchored proteins is a multi-step process involving more than 30 enzymes and can be divided into the following steps (Figure 2) 10–12:
- GPI anchor synthesis
- Protein attachment
- Lipid/glycan remodelling and protein transport
GPI anchor synthesis and protein attachment is largely carried out by a series of enzymes encoded by the PIG genes, while enzymes encoded by the PGAP genes facilitate remodeling of the GPI-anchored protein.
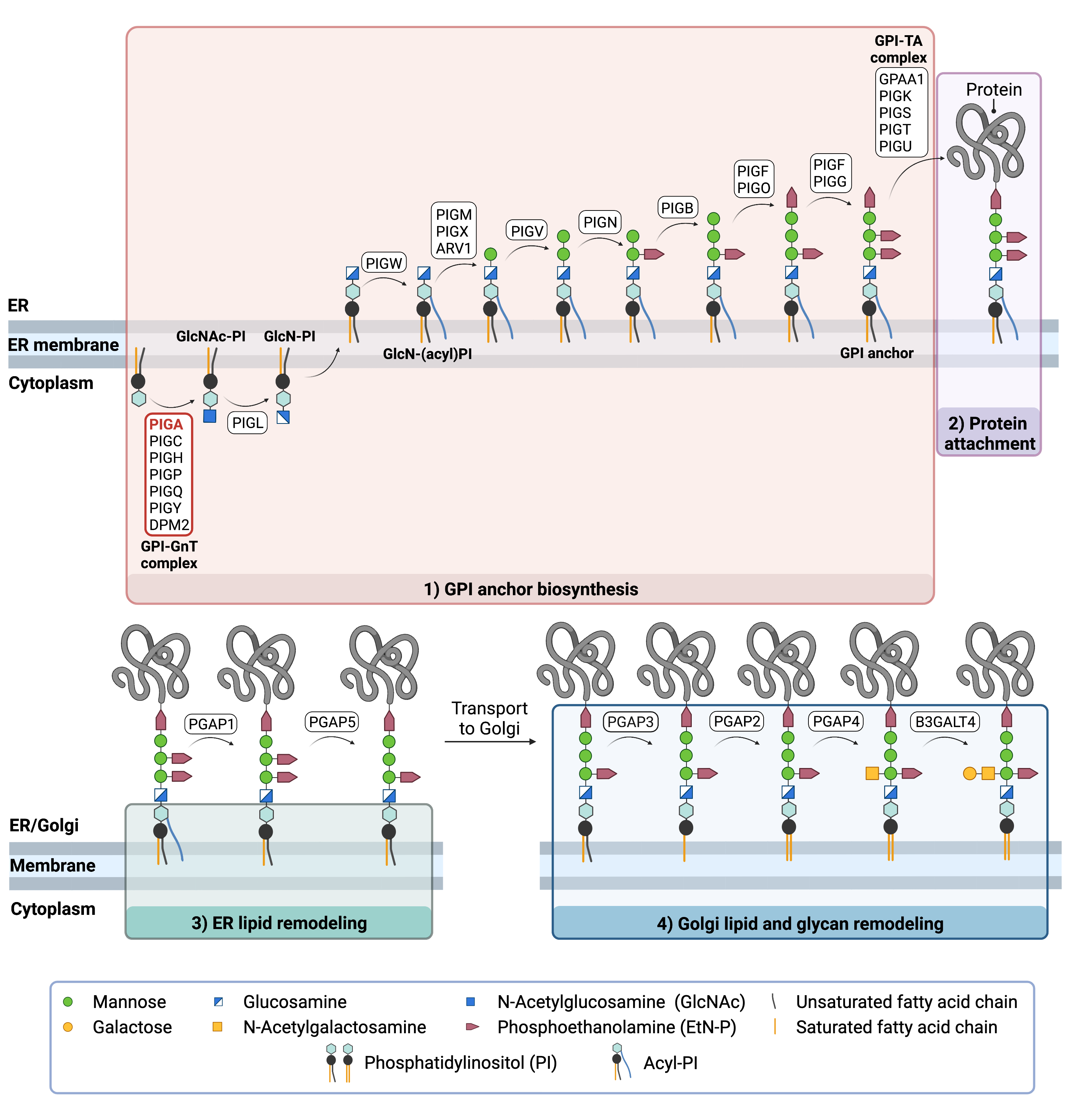
Figure 2. Overview of GPI-anchored protein biosynthesis and role of PIGA.
GPI-anchored protein biosynthesis relies on a series of enzymatic reactions. First, the GPI anchor core is built on the ER membrane, where enzymes modify the lipid and glycan portions, generating PI-GlcN-Man3-EtN-P. The protein is then attached to the GPI anchor in the ER, before further modifications are made to the lipid and glycan portions in the ER and the Golgi. PIGA is a subunit of the enzyme complex that catalyzes the first step of glycosylphosphatidylinositol (GPI) anchor protein biosynthesis.
GPI Anchor Synthesis
The first stage of GPI anchor biosynthesis involves the stepwise construction of the GPI anchor. N-acetylglucosamine (GlcNAc) is added to the lipid phosphatidylinositol (PI), generating GlcNAc-PI. The transfer of GlcNAc is catalyzed by GPI GlcNAc transferase (GPI-GnT), consisting of 7 subunits, PIG-A, PIG-C, PIG-H, PIG-P, PIG-Q, PIG-Y and DPM2—of which PIG-A is the catalytic subunit. Additional enzymes then further modify the GPI anchor structure, generating the GPI anchor core (PI-GlcN-Man3-EtN-P)11,13.
PROTEIN ATTACHMENT
Once the GPI anchor has been synthesized, it is transferred en bloc to a protein with a C-terminal GPI attachment signal sequence by GPI transamidase (GPI-TA). The five-protein enzyme complex catalyzes the simultaneous cleavage of the signal sequence and attachment of the GPI anchor to the newly synthesized protein10.
LIPID/GLYCAN REMODELING AND PROTEIN TRANSPORT
After the protein has been attached to the GPI anchor, both the glycan and lipid portion of the anchor undergo modifications (referred to as remodeling) in the ER and Golgi by post-GPI attachment to protein (PGAP) enzymes or the glycosyltransferase B3GALT4. The complete GPI-anchored protein is then transported to the plasma membrane where it associates with other GPI-anchored proteins in lipid rafts14.
Disease Mechanism
Mutations in the PIGA gene lead to loss of function of the enzyme, resulting in a failure to initiate this GPI biosynthesis pathway and a reduction in functional GPI-anchored proteins on the cell surface10–12,15. GPI-anchored proteins have several critical functions in the cell such as adhesion molecules, receptors, and enzymes in signal transduction pathways. GPI-anchored proteins are known to be expressed during neurogenesis, and even a slight deficiency in GPI-anchored proteins results in defects in neuronal development 16.
Mutations
The PIGA gene is located on the X chromosome (Xp22.2). To date, 42 variants in the PIGA gene have been reported including 34 missense variants, 4 splice site variants, and 4 truncating variants. The missense and splice site variants predispose patients to premature death— reported to be mainly due to respiratory failure2,6. Among the variants identified, c.1234C > T is the most common and most of the mutations occur in the PigA domain17.
Signs & Symptoms
Clinical Presentation
Individuals with PIGA-CDG typically develop signs and symptoms during infancy. PIGA-CDG is primarily characterized by severe to profound global developmental delay and refractory epilepsy. Symptoms of PIGA-CDG include:1,6,18–32
- Neurological – global developmental delay, intellectual disability, low muscle tone (hypotonia), epileptic seizures, and autistic features
- Dysmorphic features – facial bone abnormalities, underdeveloped nails, abnormal kidney, and genital-urinary tract features
- Dermatological – dry itchy skin (dermatitis), excess of skin, and thick and dry skin (ichthyosis)
- Gastrointestinal – feeding difficulties, stomach acid flowing back into esophagus (gastroesophageal reflux), and constipation
A less common symptom of PIGA-CDG includes heart disease, namely congenital heart disease and atrial septal defects. Other less common symptoms include vision problems, hearing problems, and liver disease.
Biochemical Abnormalities
Biochemical abnormalities observed in individuals with PIGA-CDG include elevated alkaline phosphatase levels. Alkaline phosphatase is a GPI-anchor protein that is normally found on the cell surface but may be secreted as a soluble protein in PIG deficiencies16. This has not been documented in most patients7.
Classification
Diagnosis
GPI-related CDG should be considered in individuals presenting with early onset severe seizure disorders and dysmorphic facial features, even if transferrin and total N-glycan analysis are normal33. As currently available screening tests for CDG will not reliably detect PIGA-CDG, diagnosis is typically achieved through genetic testing, either as part of an epilepsy panel or whole exome sequencing. PIGA-CDG may also be diagnosed by analyzing surface GPI-anchor proteins on blood cells by flow cytometry.
GPI-Anchored Protein Flow Cytometry
Individuals with PIGA-CDG lack GPI-anchored proteins on the surface of their granulocytes.
Biomarkers
There are no currently no known biomarkers specific to PIGA-CDG.
Prognosis
Prognosis of PIGA-CDG may vary depending on severity of an individual’s symptoms. The broad clinical spectrum may result in premature death in some patients. Age at death ranges from 15 days to 48 years of life, with half of patients passing within the first two years of life due to respiratory failure or possible sudden and unexpected death1.
Management
Management of symptoms may include combinations of physical therapy, occupational therapy, and palliative measures.
In some patients, seizures are treatable with anti-epileptic medication34. A ketogenic diet may also be used to reduce seizures1. Many children with PIGA-CDG often eventually require a feeding tube to reduce the chance of aspiration due to problems with swallowing and muscle tone abnormalities34.
Therapies
There are currently no treatment options available for PIGA-CDG, though drug repurposing, small molecule (GlcNAc-PI) and gene therapy treatments are under investigation in pre-clinical research studies3,8.
Pyridoxine, a form of vitamin B6, has been investigated as a treatment for PIGA-CDG patients with treatment-resistant epilepsy. Pyridoxine was investigated as a treatment for GPI disorders because it is thought that there may be a reduced level of intracellular pyridoxine, specifically in the brain, due to disrupted GPI-anchored protein function. Oral pyridoxine (20–30 mg/kg/day) was administered to a cohort of 4 PIGA-CDG patients for a period of 3 months and was effective in 1 patient in reducing seizure frequency35.
Research Models
Various research models are available to study PIGA-CDG, such as yeast, mice and induced pluripotent stem cells (iPSCs). A fly model and knock-in mouse model are currently being generated.
Yeast (S. cerevisiae)
Yeast gene SPT14 is homologous to human PIGA and is a member of the glycosyltransferase gene family. Spt14 mutant cells are defective in GPI anchoring due to a defect in the synthesis of GlcNAc-PI. They also display transcriptional defects and a downregulation in inositolphosphoceramide synthesis36.
Fly (D. melanogaster)
Knockout PIGA, as well as patient-specific PIGA fly models, have been generated by Dr. Clement Chow at the University of Utah School of Medicine. Phenotypic characterization is currently underway and future work will include evaluating a link between phenotypic variation and genotypic variation37.
Mouse (M. musculus)
Piga-/- constitutive knockout
Mice with germline knockout of Piga using CMV-Cre resulted in embryonic lethality with neural tube defect, edema and cleft lip/palate precluding the analysis of any postnatal neurological phenotype38.
Piga conditional-ready mice
Mutant mice strain Pigatm1Tak has exon 6 flanked by loxP sites after in vitro expression of Cre recombinase39. In studies investigating epidermal specific GPI anchor proteins, these mutant mice die 1–3 days after birth and phenotypically present with abnormal amino acid levels, impaired skin barrier function, scaly skin, and dehydration40. Other studies using targeted knockout in hematopoietic cells identified mutant mice with cleft lip/palate, edema and exencephaly41.
Pigaflox (B6.129-Pigatm1) mice were obtained from RIKEN and were previously generated by Taroh Kinoshita and Junji Takeda38.
Piga conditional central nervous system knockout mice
Piga CNS-specific conditional knockout mice do not survive past weaning and do not show structural brain defects. Mutant mice display some phenotypes observed in patients including white matter immaturity, gait imbalance, motor incoordination, and early death. Mutants also show reduced myelination and defective Purkinje cell development in the early post-natal brain42.
Pigaflox/X; Wnt1-Cre mosaic conditional knockout (cKO) mutants and Pigaflox/Y; Wnt1-Cre hemizygous cKO mutants were also generated to determine the role for GPI biosynthesis in neural crest cells (tissue-specific GPI deficiency). These mutants phenotypically showed cleft lip/cleft palate as well as craniofacial hypoplasia41.
Piga-deficient chimeric mice
Mutant mice have chimeric surface expression of GPI-anchored proteins. Chimerism of hematopoietic and non-hematopoietic tissues in mutant mice was low and GPI-cells appeared in peripheral blood of some mutant mice43.
Piga gene trap embryonic stem cell line
Mutant embryonic stem cell line PigaGt(IST13968G8)Tigm is a gene trap mutation via insertion of a gene trap vector39.
Piga knock-in mice
A knock-in mouse model is in the process of being created, beginning in 2019 by Dr. Taroh Kinoshita and Dr. Yoshiko Murakami of Osaka University37.
Human Cell Line
Induced pluripotent stem cells (iPSCs)
Hypomorphic PIGA (c.1234C>T) and PIGA-null human iPSC lines have been generated. Hypomorphic iPSCs are permissive for hematopoiesis with neuronal proliferation, differentiation, maturation, and presynaptic defects. PIGA-null iPSCs are non-permissive for hematopoiesis and differentiation16,44.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including PIGA-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Organizations
PIGA-CDG.org
GPI-anchor CDG Community Facebook Group
Multiple congenital anomalies-hypotonia-seizures syndrome type 2 (PIGA) Facebook Group
Publications
PIGA-CDG Scientific Articles on PubMed
Additional Resources
What is PIGA?
FCDGC
Kinoshita Lab (GPI anchor pathway researchers)
IEMbase
OMIM
Orphanet
GARD
Genetic Testing Registry
ClinVar
NIH
GeneCards
UniProt
References
- Johnston, J. J. et al. The phenotype of a germline mutation in PIGA: The gene somatically mutated in paroxysmal nocturnal hemoglobinuria. American Journal of Human Genetics 90, (2012).
- Bayat, A. et al. Deciphering the premature mortality in PIGA-CDG – An untold story. Epilepsy Research 170, (2021).
- FAQs - PIGA Congenital Disorder of Glycosylation (MCAHS2). https://piga-cdg.org/about-piga-cdg/role-of-piga/.
- Hill, A., Dezern, A. E., Kinoshita, T. & Brodsky, R. A. Paroxysmal nocturnal haemoglobinuria. Nat Rev Dis Primers3, (2017).
- Brodsky, R. A. Paroxysmal nocturnal hemoglobinuria. Blood 124, 2804–2811 (2014).
- Bayat, A. et al. Lessons learned from 40 novel PIGA patients and a review of the literature. Epilepsia 61, (2020).
- Bellai-Dussault, K., Nguyen, T. T. M., Baratang, N. V., Jimenez-Cruz, D. A. & Campeau, P. M. Clinical variability in inherited glycosylphosphatidylinositol deficiency disorders. Clinical Genetics vol. 95 (2019).
- Home | World CDG Organization. https://worldcdg.org/.
- Kinoshita, T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biology 10, 190290 (2020).
- Kinoshita, T. Glycosylphosphatidylinositol (GPI) anchors: Biochemistry and cell biology: Introduction to a thematic review series. Journal of Lipid Research vol. 57 (2016).
- Liu, Y. S. & Fujita, M. Mammalian GPI-anchor modifications and the enzymes involved. Biochemical Society Transactions vol. 48 (2020).
- Englund, P. T. The Structure and Biosynthesis of Glycosyl Phosphatidylinositol Protein Anchors. Annual Review of Biochemistry 62, (1993).
- Maeda, Y. & Kinoshita, T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Progress in Lipid Research vol. 50 (2011).
- Fujita, M. & Kinoshita, T. Structural remodeling of GPI anchors during biosynthesis and after attachment to proteins.FEBS Letters vol. 584 (2010).
- Murakami, Y. & Kinoshita, T. Congenital Disorders of Glycosylation: Glycosylphosphatidylinositol (GPI)-Related. Glycoscience: Biology and Medicine 1229–1236 (2015) doi:10.1007/978-4-431-54841-6_171.
- Yuan, X. et al. A hypomorphic PIGA gene mutation causes severe defects in neuron development and susceptibility to complement-mediated toxicity in a human iPSC model. PLoS ONE 12, (2017).
- Wu, T. et al. The Glycosylphosphatidylinositol biosynthesis pathway in human diseases. Orphanet Journal of Rare Diseases 15, 1–11 (2020).
- Belet, S. et al. Early frameshift mutation in PIGA identified in a large XLID family without neonatal lethality. Human Mutation 35, (2014).
- Kato, M. et al. PIGA mutations cause early-onset epileptic encephalopathies and distinctive features. Neurology 82, (2014).
- Kim, Y. O. et al. A novel PIGA mutation in a family with X-linked, early-onset epileptic encephalopathy. Brain and Development 38, (2016).
- Joshi, C. et al. Ketogenic diet – A novel treatment for early epileptic encephalopathy due to PIGA deficiency. Brain and Development 38, (2016).
- Fauth, C. et al. A recurrent germline mutation in the PIGA gene causes Simpson-Golabi-Behmel syndrome type 2. American Journal of Medical Genetics, Part A 170, (2016).
- van der Crabben, S. N. et al. Expanding the spectrum of phenotypes associated with germline PIGA mutations: A child with developmental delay, accelerated linear growth, facial dysmorphisms, elevated alkaline phosphatase, and progressive CNS abnormalities. American Journal of Medical Genetics, Part A 164, (2014).
- Swoboda, K. J. et al. A novel germline PIGA mutation in Ferro-Cerebro-Cutaneous syndrome: A neurodegenerative X-linked epileptic encephalopathy with systemic iron-overload. American Journal of Medical Genetics, Part A 164, (2014).
- Tarailo-Graovac, M. et al. The genotypic and phenotypic spectrum of PIGA deficiency. Orphanet Journal of Rare Diseases 10, (2015).
- Xie, L. ling, Song, X. jie, Li, T. yi & Jiang, L. A novel germline PIGA mutation causes early-onset epileptic encephalopathies in Chinese monozygotic twins. Brain and Development 40, (2018).
- Low, K. J. et al. A novel PIGA variant associated with severe X-linked epilepsy and profound developmental delay. Seizure 56, (2018).
- Lin, W. De, Chou, I. C., Tsai, F. J. & Hong, S. Y. A novel PIGA mutation in a Taiwanese family with early-onset epileptic encephalopathy. Seizure 58, (2018).
- Yang, J. et al. A likely pathogenic variant putatively affecting splicing of PIGA identified in a multiple congenital anomalies hypotonia-seizures syndrome 2 (MCAHS2) family pedigree via whole-exome sequencing. Molecular Genetics and Genomic Medicine 6, (2018).
- Cash, S. J., Mcgue, B. P., Reynolds, T. S. & Crist, E. R. PIGA related disorder as a range of phenotypes rather than two distinct subtypes. Brain and Development 42, (2020).
- Neuhofer, C. M. et al. A Novel Mutation in PIGA Associated with Multiple Congenital Anomalies-Hypotonia-Seizure Syndrome 2 (MCAHS2) in a Boy with a Combination of Severe Epilepsy and Gingival Hyperplasia. Molecular Syndromology 11, (2020).
- Jiao, X. et al. Analyzing clinical and genetic characteristics of a cohort with multiple congenital anomalies-hypotonia-seizures syndrome (MCAHS). Orphanet Journal of Rare Diseases 15, (2020).
- Lam, C. et al. Expanding the clinical and molecular characteristics of PIGT-CDG, a disorder of glycosylphosphatidylinositol anchors. Molecular Genetics and Metabolism 115, 128–140 (2015).
- PIGA-congenital disorder of glycosylation (PIGA-CDG, also known as phosphatidylinositol-glycan class A protein deficiency or Multiple Congenital Anomalies-Hypotonia-Seizures Syndrome 2). It belongs to a group of disorders called Glycosylphosphatidylinosi | Rare Diseases Clinical Research Network. https://www.rarediseasesnetwork.org/fcdgc/piga.
- Bayat, A. et al. Pyridoxine or pyridoxal-5-phosphate treatment for seizures in glycosylphosphatidylinositol deficiency: A cohort study. Developmental Medicine & Child Neurology (2022).
- Schönbächler, M., Horvath, A., Fassler, J. & Riezman, H. The yeast spt14 gene is homologous to the human PIG-A gene and is required for GPI anchor synthesis. EMBO J 14, 1637–45 (1995).
- PIGA Research Projects - PIGA Congenital Disorder of Glycosylation (MCAHS2). https://piga-cdg.org/research/current-research-projects/.
- Nozaki, M. et al. Developmental abnormalities of glycosylphosphatidylinositol-anchor-deficient embryos revealed by Cre/loxP system. Lab Invest 79, 293–9 (1999).
- All Phenotypes Piga MGI Mouse. http://www.informatics.jax.org/allele/allgenoviews/MGI:2386120.
- Hara-Chikuma, M. et al. Epidermal-Specific Defect of GPI Anchor in Pig-a Null Mice Results in Harlequin Ichthyosis-Like Features. Journal of Investigative Dermatology 123, 464–469 (2004).
- Lukacs, M., Roberts, T., Chatuverdi, P. & Stottmann, R. W. Glycosylphosphatidylinositol biosynthesis and remodeling are required for neural tube closure, heart development, and cranial neural crest cell survival. Elife 8, (2019).
- Lukacs, M., Blizzard, L. E. & Stottmann, R. W. CNS glycosylphosphatidylinositol deficiency results in delayed white matter development, ataxia and premature death in a novel mouse model. Human Molecular Genetics 29, 1205–1217 (2020).
- Brasil, S. et al. CDG therapies: From bench to bedside. International Journal of Molecular Sciences vol. 19 (2018).
- Yuan, X. et al. Generation of Glycosylphosphatidylinositol Anchor Protein-Deficient Blood Cells From Human Induced Pluripotent Stem Cells. Stem Cells Translational Medicine 2, 819–829 (2013).