
Lay Summary
PGAP3-CDG, also known as hyperphosphatasia with mental retardation syndrome 4 (HPMRS4) or Mabry syndrome, is a rare inherited neurological disorder. To date, more than 30 cases of PGAP3-CDG have been reported in the medical literature. PGAP3-CDG is classified as a disorder of GPI anchor biosynthesis. PGAP3-CDG is caused when an individual has mutations in both copies of their PGAP3 gene, which provides instructions for making a protein that participates in building GPI anchors. GPI anchors are molecules that “anchor” certain proteins to the cell surface. The PGAP3 protein functions in the final steps of GPI anchor synthesis in the Golgi, which involves modifying the lipid (fat) component of the GPI anchor. These modifications to the lipid are needed for GPI-anchored proteins to be able to attach to the cell surface. Mutations in the PGAP3 gene cause GPI-anchored proteins to be unstable or unable to attach to the cell surface. Symptoms of PGAP3-CDG begin in infancy and are primarily characterized by developmental delay, intellectual disability, low muscle tone, epilepsy, and increased alkaline phosphatase in the blood (hyperphosphatasia). Abnormal facial features and behavioural symptoms are also common. Screening tests are available for PGAP3-CDG, but a definitive diagnosis is achieved through genetic testing. There are currently no approved treatments for PGAP3-CDG. Treatment is focused on the management of specific symptoms and preventing complications.
Overview
Post-GPI attachment to proteins phospholipase 3 congenital disorder of glycosylation (PGAP3-CDG) is a rare autosomal recessive genetic disorder. The first reported case of PGAP3-CDG was in 2012 and there are over 30 confirmed cases to date1–9. PGAP3-CDG is commonly referred to as hyperphosphatasia with mental retardation syndrome 4 (HPMRS4) or Mabry syndrome.
The PGAP3 gene encodes an enzyme (PGAP3) that is involved in the final steps of glycosylphosphatidylinositol (GPI)-anchored protein biosynthesis. PGAP3 participates in lipid remodelling of GPI-anchored proteins in the Golgi by removing an unsaturated fatty acid chain so that it can be replaced by a saturated chain. Lipid remodelling enables GPI-anchored proteins to associate with lipid rafts in the plasma membrane which play an important role in cell signalling. Deficiency in the PGAP3 enzyme result in GPI-anchored proteins being unable to associate with lipid rafts, which has downstream effects on protein trafficking and sorting10–12.
Symptoms of PGAP3-CDG begin at infancy and during the first years of life, and the characteristic presentation includes developmental delay, intellectual disability, low muscle tone, epilepsy, increased alkaline phosphatase in the blood (hyperphosphatasia), abnormal facial features, and behaviour symptoms, such as autistic behaviour and sleep disturbances13. Although a definitive diagnosis can only be achieved by genetic sequencing, analyzing blood cells for an absence of GPI-anchored proteins can help diagnose disorders of GPI-anchor biosynthesis. There are currently no approved treatments for PGAP3-CDG13.
Synonyms
- Post-GPI attachment to proteins phospholipase 3 congenital disorder of glycosylation
- Proteins phospholipase 3 deficiency
- Hyperphosphatasia with mental retardation 4 (HPMRS4)
- Mabry syndrome
Inheritance
PGAP3-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The PGAP3 gene encodes a GPI-specific phospholipase (PGAP3) that participates in lipid remodelling of GPI-anchored proteins in the Golgi. PGAP3 removes the unsaturated fatty acid chain from GPI so that it can be replaced by a saturated chain. Lipid remodelling is critical for GPI-anchored proteins to associate in lipid rafts in the plasma membrane. Lipid rafts are domains within the plasma membrane that are enriched in glycosphingolipids, cholesterol, and GPI-anchored proteins and regulate different cellular processes, such as signalling transduction10–12.
GPI-Anchored Protein Biosynthesis
GPI-anchor biosynthesis is one of the major glycosylation pathways that attach glycans to lipid molecules within cells. Many proteins, called GPI-anchored proteins are attached to the cell surface by GPI anchors.
The core structure of GPI consists of phosphatidylinositol (PI), glucosamine (GlcN), three mannose sugars (Man3), and phosphoethanolamine (EtN-P) connected to each other in that sequence (Figure 1). GPI-anchored proteins are attached to the GPI by forming a bond between the EtN-P group of the GPI core and the C-terminus of newly synthesized proteins in the ER lumen14.
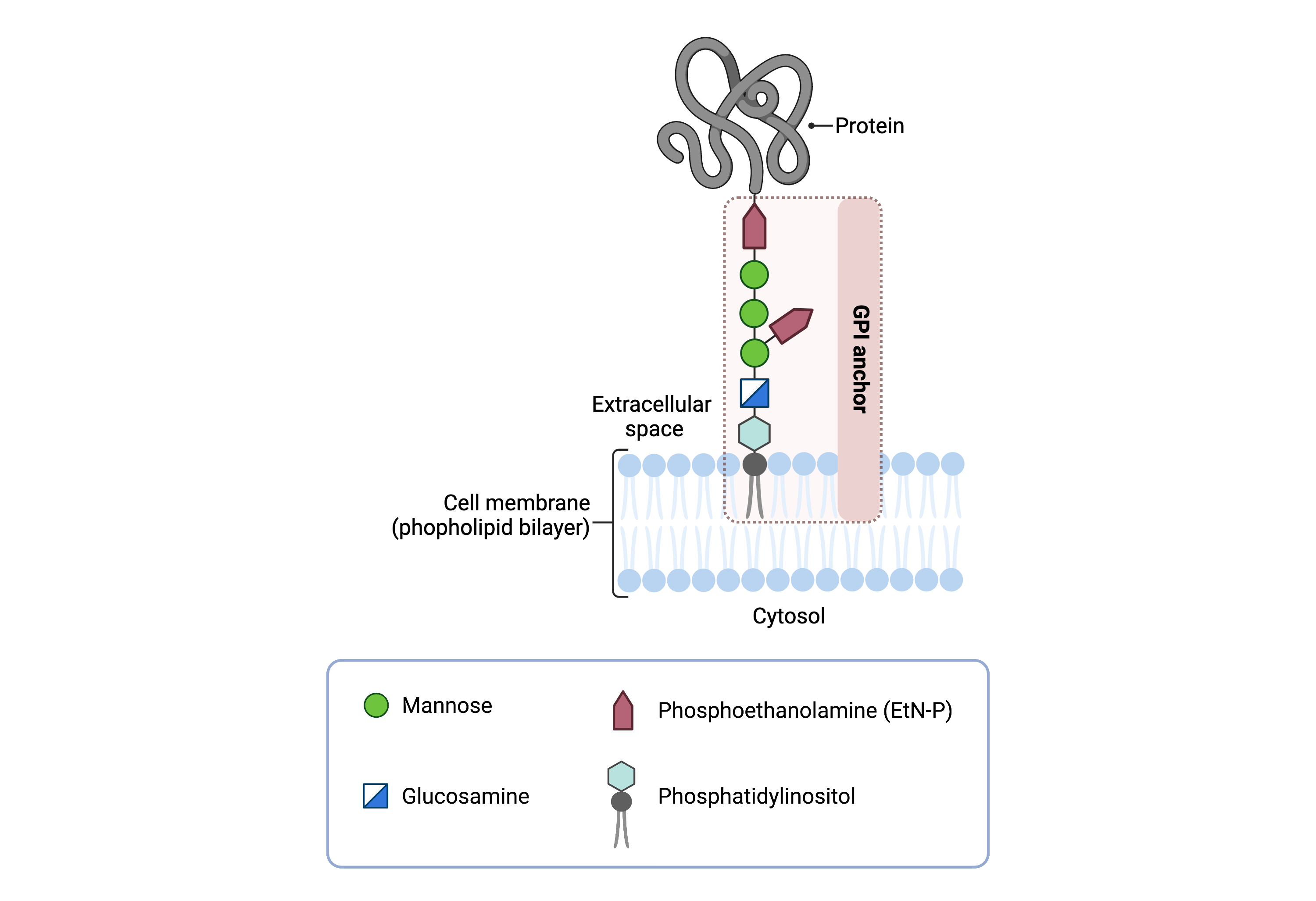
Figure 1. Overview of GPI-anchored protein structure.
The GPI anchor is added to proteins, generating GPI-anchored proteins. The GPI anchor section lodges into the membrane, attaching the protein to the membrane. The GPI anchor involves phosphatidylinositol, glucosamine, mannose and phosphoethanolamine.
The generation of a GPI-anchored proteins is a multi-step process involving more than 30 enzymes and can be divided into the following steps15–17:
- GPI anchor synthesis
- Protein attachment
- Lipid /glycan remodelling and protein transport
GPI-anchored protein biosynthesis and protein attachment is largely carried out by a series of enzymes encoded by the PIG genes, while enzymes encoded by the PGAP genes facilitate remodelling of the GPI anchored protein (Figure 2).
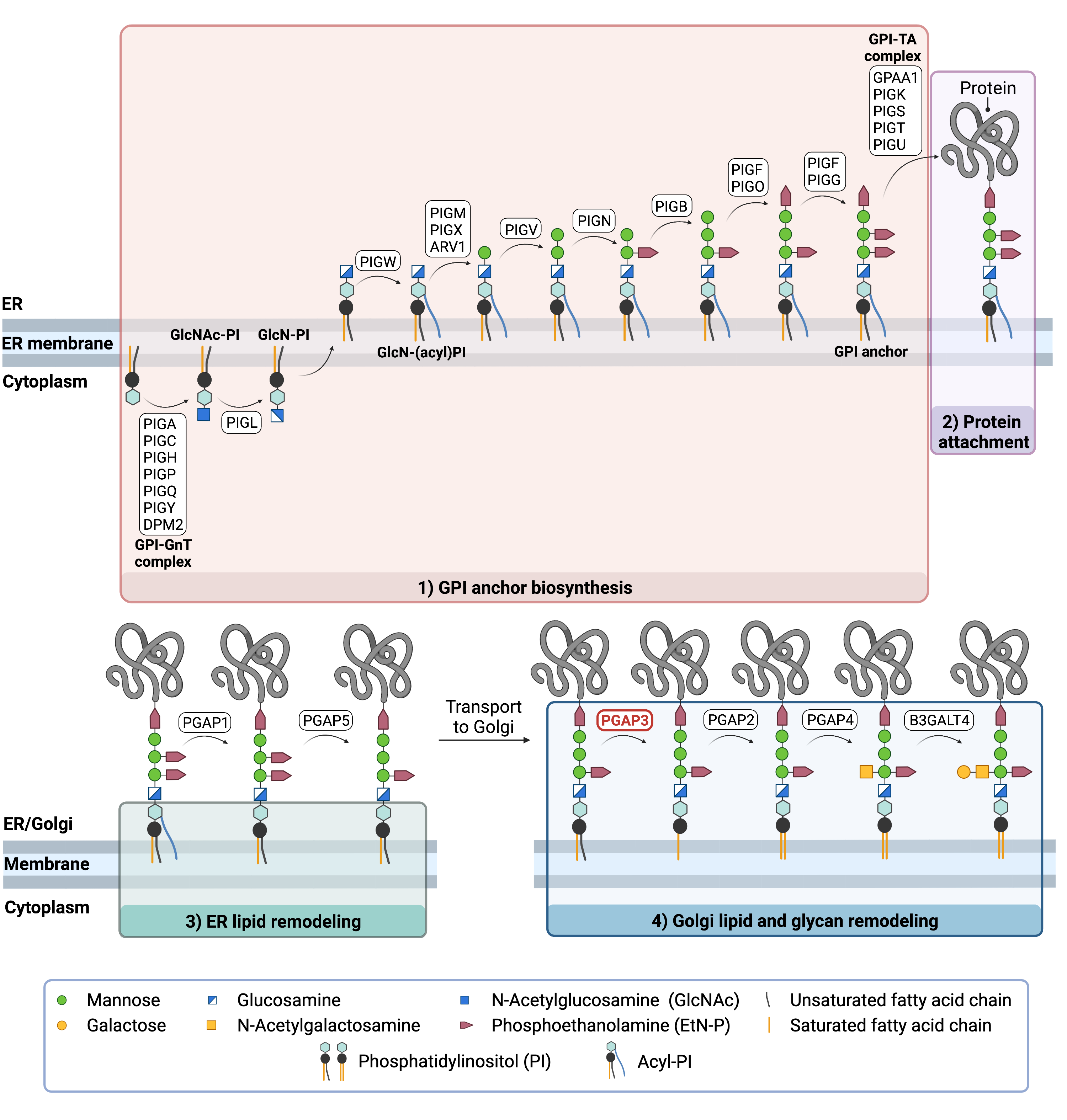
Figure 2. Overview of GPI-anchored protein biosynthesis and role of PGAP3.
GPI-anchored protein biosynthesis relies on a series of enzymatic reactions. First, the GPI anchor core is built on the ER membrane, where enzymes modify the lipid and glycan portions, generating PI-GlcN-Man3-EtN-P. The protein is then attached to the GPI anchor in the ER, before further modifications are made to the lipid and glycan portions in the ER and the Golgi. PGAP3 is an enzyme that is involved in the final steps of GPI-anchored protein biosynthesis, where PGAP3 removes an unsaturated fatty acid chain from the GPI-anchored protein so that it can be replaced by a saturated chain.
GPI ANCHOR SYNTHESIS
The first stage of GPI anchor biosynthesis involves the stepwise construction of the GPI anchor. N-acetylglucosamine (GlcNAc) is added to the lipid phosphatidylinositol (PI), generating GlcNAc-PI. The transfer of GlcNAc is catalyzed by GPI GlcNAc transferase (GPI-GnT). Additional enzymes then further modify the GPI anchor structure, generating the GPI anchor core (PI-GlcN-Man3-EtN-P)16,19.
PROTEIN ATTACHMENT
Once the GPI anchor has been synthesized, it is transferred "en bloc" to a protein with a C-terminal GPI attachment signal sequence by the GPI transamidase (GPI-TA) complex. This complex catalyzes the simultaneous cleavage of the signal sequence and attachment of the GPI anchor to the newly synthesized protein15.
LIPID/GLYCAN REMODELING AND PROTEIN TRANSPORT
After the protein has been attached to the GPI anchor, both the glycan and lipid portion of the anchor undergo modifications (referred to as remodeling) in the ER and Golgi by post-GPI attachment to protein (PGAP) enzymes or the glycosyltransferase B3GALT4; PGAP3 catalyzes the removal of the unsaturated fatty acid chain in the Golgi10,12. The complete GPI-anchored protein is then transported to the plasma membrane where it associates with other GPI-anchored proteins in lipid rafts11,12.
Disease Mechanism
Mutations to the PGAP3 gene lead to a reduction in functional GPI-anchored proteins, as PGAP3 is required for remodelling of GPI-anchored proteins. Remodelling is required for proper association between GPI-anchored proteins and lipid rafts, which impacts trafficking and sorting of proteins10,11,21. Elevated levels of alkaline phosphatase in the blood (hyperphosphatasia) is common in patients with PGAP3-CDG and is a result of GPI-anchored protein release from the cell surface9. Additionally, PGAP3 has been shown to be essential to early brain development and mutations lead to phenotypes such as seizures and hypotonia3.
Mutations
The PGAP3 gene is located on chromosome 17 (17q12). Exons 3 and 7 appear to be mutated most frequently, with the majority of mutations being missense mutations. Frameshift, splice site and 3’UTR mutations have also been reported1.
Signs & Symptoms
Clinical Presentation
Individuals with PGAP3-CDG typically develop signs and symptoms during infancy or in the first years of life. PGAP3-CDG is primarily characterized by neurological symptoms, abnormal facial features and increased levels of alkaline phosphatase in the blood. Common symptoms of PGAP3-CDG include1–9,13,22,23:
- Neurological – severe global, speech and language developmental delays, intellectual disability, low muscle tone (hypotonia), lack of muscle control and coordination (ataxia), and epileptic seizures
- Dysmorphic features – distinct facial abnormalities, such as a broad nasal bridge and tip, short nose, long palpebral fissures, increased distance between the eyes, long philtrum, thin and wide upper lip, large ear lobes, and full cheeks
- Behavioural – autistic behaviour and sleep disturbances
Less common symptoms of PGAP3-CDG include a cleft palate and a small head (microcephaly), decreased intrauterine fetal movements, cardiac problems, abnormal corpus callosum, and olfactory bulb agenesis.
Biochemical Presentation
Elevated levels of alkaline phosphatase in the blood (hyperphosphatasia) are often observed in individuals with PGAP3-CDG13,22.
Classification
PGAP3-CDG is classified as a disorder of GPI anchor biosynthesis.
Diagnosis
GPI-related CDG should be considered in individuals presenting with early onset severe seizure disorders and dysmorphic facial features, even if transferrin and total N-glycan analysis are normal. As currently available screening tests for CDG will not reliably detect PGAP3-CDG, diagnosis is typically achieved through genetic testing, either as part of an epilepsy panel or whole exome sequencing. PGAP3-CDG may also be primarily screened for by analyzing surface GPI-anchor proteins on blood cells by flow cytometry9,13.
GPI-Anchored Protein Flow Cytometry
Individuals with PGAP3-CDG show an absence of GPI-anchored proteins on the surface of granulocytes, a type of white blood cell9,13.
Biomarkers
No biomarkers for PGAP3-CDG have been reported.
Prognosis
Prognosis of PGAP3-CDG may vary depending on the severity of an individual’s symptoms. With the majority of PGAP3-CDG patients reported in the medical literature being young, the long-term prognosis is difficult to determine13.
Management
Management of symptoms may include combinations of physical therapy, occupational therapy, and palliative measures. In some patients, seizures are treatable with anti-epileptic medication, such as pyridoxine (vitamin B6)13,24.
Therapies
There are currently no treatment options available for PGAP3-CDG, although several therapies are currently under investigation by Moonshots for Unicorns including gene therapy and drug repurposing.
Research Models
Several PGAP3 research models have been generated including in yeast, zebrafish, mice, and mouse cell lines.
Yeast (S. cerevisiae)
PGAP3 homologue in yeast Per1, is required for production of lyso-GPI, however, it is localized to the ER in yeast rather than the Golgi. Similar to PGAP3, Per1 is essential for fatty acid remodeling and association of GPI anchor proteins with lipid rafts. Yeast deficient in per1 and gup1 (per1Δ and gup1Δ) have significantly delayed transport of GPI-anchored proteins12.
Fish (D. rerio)
Knockdown of Pgap3 in zebrafish impacts early brain development and results in neural tube defects, delayed development, and altered neuronal pathways in the brain and spinal cord, which led to a seizure-like phenotype. Mutant zebrafish also displayed dysmorphic features, such as a bent head and low-set ears3.
Chinese Hamster Ovary Cell Line
Chinese hamster ovary (CHO) cells with mutations in both PGAP3 and PGAP2 genes (referred to as double mutants) were used by Maeda et al. (2007) to determine the significance of fatty acid remodeling of GPI anchor proteins on lipid raft association. Double mutants had unsaturated chains in the sn-2 position on the GPI anchor protein, compared to wild-type CHO cells which had a saturated fatty acid chain, suggesting PGAP2 and PGAP3 were involved in lipid remodelling. And PGAP3- and PGAP2-mediated remodelling was found to be essential for GPI-anchored proteins to be incorporated into lipid rafts11.
Mouse (M. musculus)
Conditional knockout embryonic stem cell line
Pgap3tm1a(KOMP)Wtsi and Pgap3tm2a(KOMP)Wtsi are mice embryonic stem cell lines with targeted null/knockout mutations of the first allele of Pgap3 (IMSR).
Pgap3-/- knockout mouse
Pgap3-/- mice developed autoimmune-like symptoms, such as spontaneous germinal center formation and enlarged renal glomeruli. These mice also did not have fatty acid remodeling of GPI-anchored proteins. Pgap3-/- mice were used to demonstrate that PGAP3-dependent fatty acid remodeling of GPI-anchored proteins have a role in autoimmunity. Mice with targeted mutations in either B or T cells did not develop autoimmune-like symptoms25.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including PGAP3-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Organizations/Groups
GPI-anchor CDG Community Facebook Group
Mabry Syndrome Awareness Facebook Group
Publications
PGAP3-CDG Scientific Articles on PubMed
Additional Resources
Kinoshita Lab (GPI anchor pathway researchers)
PGAP3-CDG on FCDGC
Mabry syndrome, Medline Plus
IEMbase
OMIM
Orphanet
Genetic Testing Registry
ClinVar
NIH
GeneCards
UniProt
References
- Abi Farraj, L. et al. Clinical, genetic, and molecular characterization of hyperphosphatasia with mental retardation: A case report and literature review. Diagnostic Pathology vol. 14 (2019).
- Akgün Doğan, Ö. et al. Hyperphosphatasia with mental retardation syndrome type 4 In two siblings-expanding the phenotypic and mutational spectrum. European Journal of Medical Genetics 62, (2019).
- Da’as, S. I. et al. PGAP3 Associated with Hyperphosphatasia with Mental Retardation Plays a Novel Role in Brain Morphogenesis and Neuronal Wiring at Early Development. Cells 9, (2020).
- Bezuidenhout, H. et al. Hyperphosphatasia with mental retardation syndrome type 4 in three unrelated South African patients. American Journal of Medical Genetics, Part A 182, 2230–2235 (2020).
- Abdel-Hamid, M. S. et al. PGAP3-related hyperphosphatasia with mental retardation syndrome: Report of 10 new patients and a homozygous founder mutation. Clinical genetics 93, 84–91 (2018).
- Thompson, M. D. et al. Phenotypic variability in hyperphosphatasia with seizures and neurologic deficit (Mabry syndrome). American journal of medical genetics. Part A 158A, 553–558 (2012).
- Akgün Doğan, Ö. et al. Hyperphosphatasia with mental retardation syndrome type 4 In two siblings-expanding the phenotypic and mutational spectrum. European journal of medical genetics 62, (2019).
- Balobaid, A. et al. Delineating the phenotypic spectrum of hyperphosphatasia with mental retardation syndrome 4 in 14 patients of Middle-Eastern origin. American journal of medical genetics. Part A 176, 2850–2857 (2018).
- Howard, M. F. et al. Mutations in PGAP3 impair GPI-anchor maturation, causing a subtype of hyperphosphatasia with mental retardation. American Journal of Human Genetics 94, 278–287 (2014).
- Kinoshita, T. Biosynthesis and deficiencies of glycosylphosphatidylinositol. Proceedings of the Japan Academy. Series B, Physical and Biological Sciences 90, 130 (2014).
- Maeda, Y. et al. Fatty Acid Remodeling of GPI-anchored Proteins Is Required for Their Raft Association. Molecular Biology of the Cell 18, 1497 (2007).
- Maeda, Y. & Kinoshita, T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Progress in Lipid Research 50, 411–424 (2011).
- PGAP3 Congenital Disorder of Glycosylation | Rare Diseases Clinical Research Network. https://www.rarediseasesnetwork.org/fcdgc/pgap3.
- Kinoshita, T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biology 10, 190290 (2020).
- Kinoshita, T. Glycosylphosphatidylinositol (GPI) anchors: Biochemistry and cell biology: Introduction to a thematic review series. Journal of Lipid Research vol. 57 (2016).
- Liu, Y. S. & Fujita, M. Mammalian GPI-anchor modifications and the enzymes involved. Biochemical Society Transactions vol. 48 (2020).
- Englund, P. T. The Structure and Biosynthesis of Glycosyl Phosphatidylinositol Protein Anchors. Annual Review of Biochemistry 62, (1993).
- Bayat, A. et al. Deciphering the premature mortality in PIGA-CDG – An untold story. Epilepsy Research 170, (2021).
- Maeda, Y. & Kinoshita, T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Progress in Lipid Research vol. 50 (2011).
- Fujita, M. & Kinoshita, T. Structural remodeling of GPI anchors during biosynthesis and after attachment to proteins.FEBS Letters vol. 584 (2010).
- Maeda, Y. et al. Fatty Acid Remodeling of GPI-anchored Proteins Is Required for Their Raft Association □ D.Molecular Biology of the Cell 18, 1497–1506 (2007).
- Wu, T. et al. The Glycosylphosphatidylinositol biosynthesis pathway in human diseases. Orphanet Journal of Rare Diseases 15, 1–11 (2020).
- Knaus, A. et al. Rare Noncoding Mutations Extend the Mutational Spectrum in the PGAP3 Subtype of Hyperphosphatasia with Mental Retardation Syndrome. Human Mutation 37, 737–744 (2016).
- Sakaguchi, T. et al. A novel PGAP3 mutation in a Croatian boy with brachytelephalangy and a thin corpus callosum. Nature Publishing Group 5, (2018).
- Wang, Y. et al. Significance of Glycosylphosphatidylinositol-anchored Protein Enrichment in Lipid Rafts for the Control of Autoimmunity. The Journal of Biological Chemistry 288, 25490 (2013).