
Lay Summary
PIGG-CDG, also known as PIGG deficiency, is a rare inherited condition that predominantly affects the nervous system. Almost 30 cases of PIGG-CDG have been reported in the literature to date. PIGG-CDG is classified as a disorder of GPI anchor biosynthesis. PIGG-CDG is caused when an individual has a mutation in both copies of their PIGG gene. The PIGG gene provides instructions for making a protein that participates in building GPI anchors - molecules that “anchor” certain proteins to the cell surface. The PIG-G enzyme is involved in transferring a molecule called ethanolamine phosphate to a specific mannose sugar on the GPI-anchor while it is being built in the endoplasmic reticulum (ER). Mutations in the PIGG gene cause defects in GPI-anchor proteins which make them unstable or unable to attach to the cell surface. Symptoms of PIGG-CDG being at infancy and are primarily characterized by developmental delay, intellectual disability, poor muscle tone and seizures. PIGG-CDG is usually diagnosed through genetic testing, however testing for the presence of GPI-anchor proteins on certain blood cells can also identify PIGG-CDG. There are currently no approved treatments for PIGG-CDG, however there are medications and supplements available that may aid in the management of specific symptoms.
Overview
Phosphatidylinositol Glycan Anchor Biosynthesis class G congenital disorder of glycosylation (PIGG-CDG) is a rare autosomal recessive genetic disorder. The first reported case was in 2016 and 291–3 confirmed cases have been reported in the medical literature to date.
The PIGG gene encodes the catalytic component of a complex that transfers ethanolamine phosphate to the second mannose residue on the glycosylphosphatidylinositol (GPI) anchor during GPI-anchor protein biosynthesis 2,4. GPI anchors are an important mechanism of attachment for proteins to the cell surface, and these proteins are known as GPI-anchored proteins. Mutations in PIGG result in a reduction of functional GPI-anchor proteins on the cell surface, where they are critical to development and function of many organs and the nervous system 2,4.
Symptoms begin at infancy and the characteristic presentation includes developmental delay, intellectual disability, seizures and hypotonia. Although a definitive diagnosis can only be achieved through genetic sequencing, analyzing blood cells and fibroblast cells for an absence of GPI-anchored proteins can help diagnose disorders of GPI-anchor biosynthesis 1–3. No treatment is currently available for PIGG-CDG although medication and supplements, such as pyridoxine, may be used to treat specific symptoms 2.
Synonyms
- Phosphatidylinositol glycan class G congenital disorder of glycosylation
- PIGG deficiency
- Glycosylphosphatidylinositol Biosynthesis Defect 13 (GPIBD13)
- Intellectual Developmental Disorder, Autosomal Recessive 53
- Mental retardation, autosomal recessive 53
Inheritance
PIGG-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The Phosphatidylinositol Glycan Anchor Biosynthesis, class G (PIG-G) gene encodes the PIG-G protein that forms a complex with PIG-F to form the catalytic component of an ethanolamine phosphate transferase enzyme (GPI-EtNP transferase II)5. PIG-F stabilizes PIG-G in the complex, and PIG-G competes with PIG-O for the binding site of PIG-F. The GPI-EtNP transferase II enzyme is located in the endoplasmic reticulum (ER) and is involved in the addition of one ethanolamine phosphate (EtNP) group to the second mannose residue of glycosylpohsphatidylinosital (GPI) during GPI-anchor protein biosynthesis2,4.
GPI-Anchored Protein Biosynthesis
GPI-anchored protein biosynthesis is one of the major glycosylation pathways that attach glycans to lipid molecules within cells. Many proteins are attached to the cell surface by GPI anchors, which are referred to as GPI-anchored proteins.
The core structure of GPI consists of phosphatidylinositol (PI), glucosamine (GlcN), three mannose sugars (Man3), and phosphoethanolamine (EtN-P) connected to each other in that sequence (Figure 1). GPI-anchored proteins are attached to the GPI by forming a bond between the EtN-P group of the GPI core and the C-terminus of newly synthesized proteins in the ER lumen6.
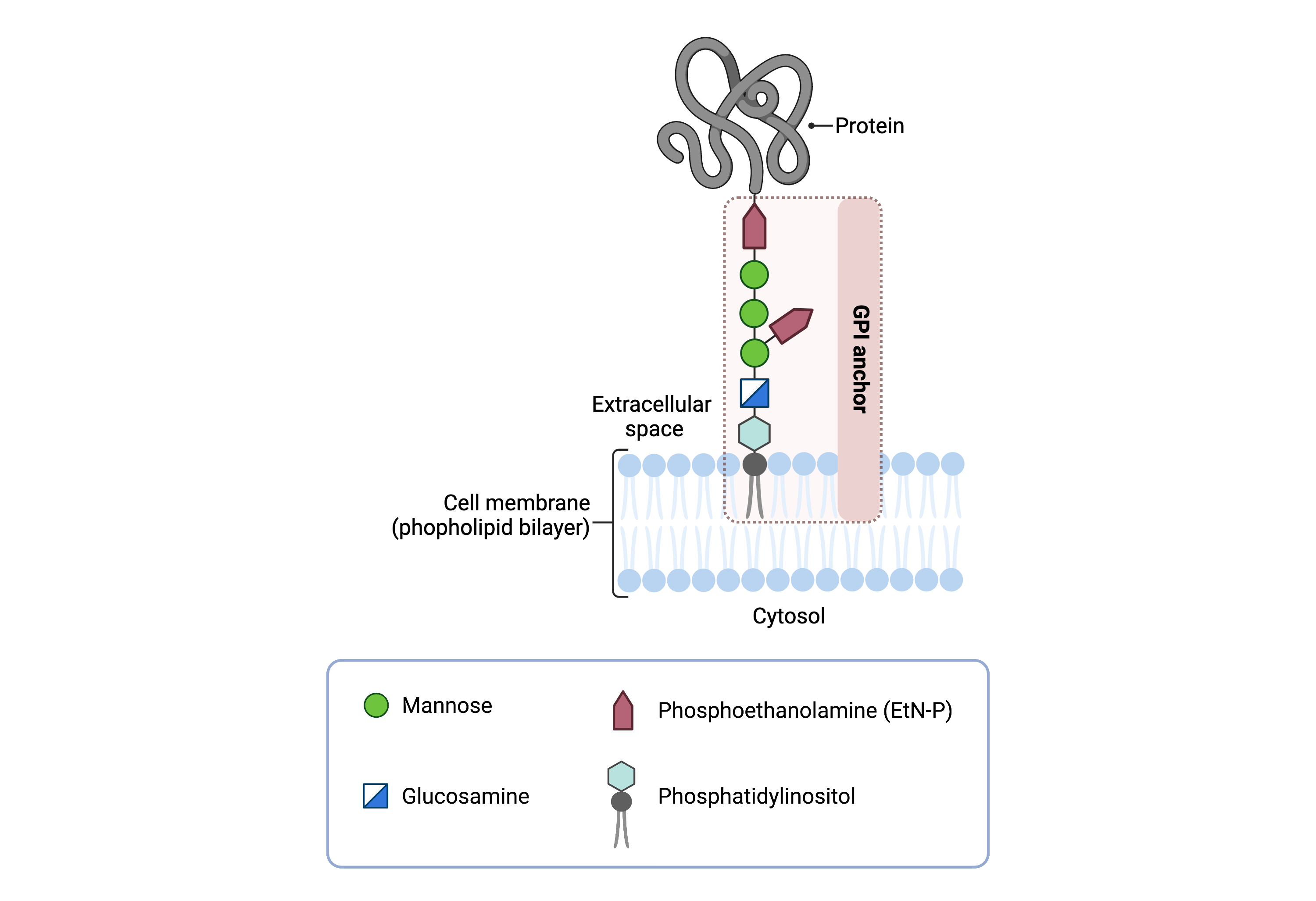
Figure 1: Overview of GPI-anchored protein structure.
The GPI anchor is added to proteins, generating GPI-anchored proteins. The GPI anchor section lodges into the membrane, attaching the protein to the membrane. The GPI anchor involves phosphatidylinositol, glucosamine, mannose and phosphoethanolamine.
The generation of GPI-anchored proteins is a multi-step process involving more than 30 enzymes and can be divided into the following steps (Figure 2) 7–9:
- GPI anchor synthesis
- Protein attachment
- Lipid/glycan remodelling and protein transport
GPI anchor synthesis and protein attachment is largely carried out by a series of enzymes encoded by the PIG genes, while enzymes encoded by the PGAP genes facilitate remodeling of the GPI-anchored protein.
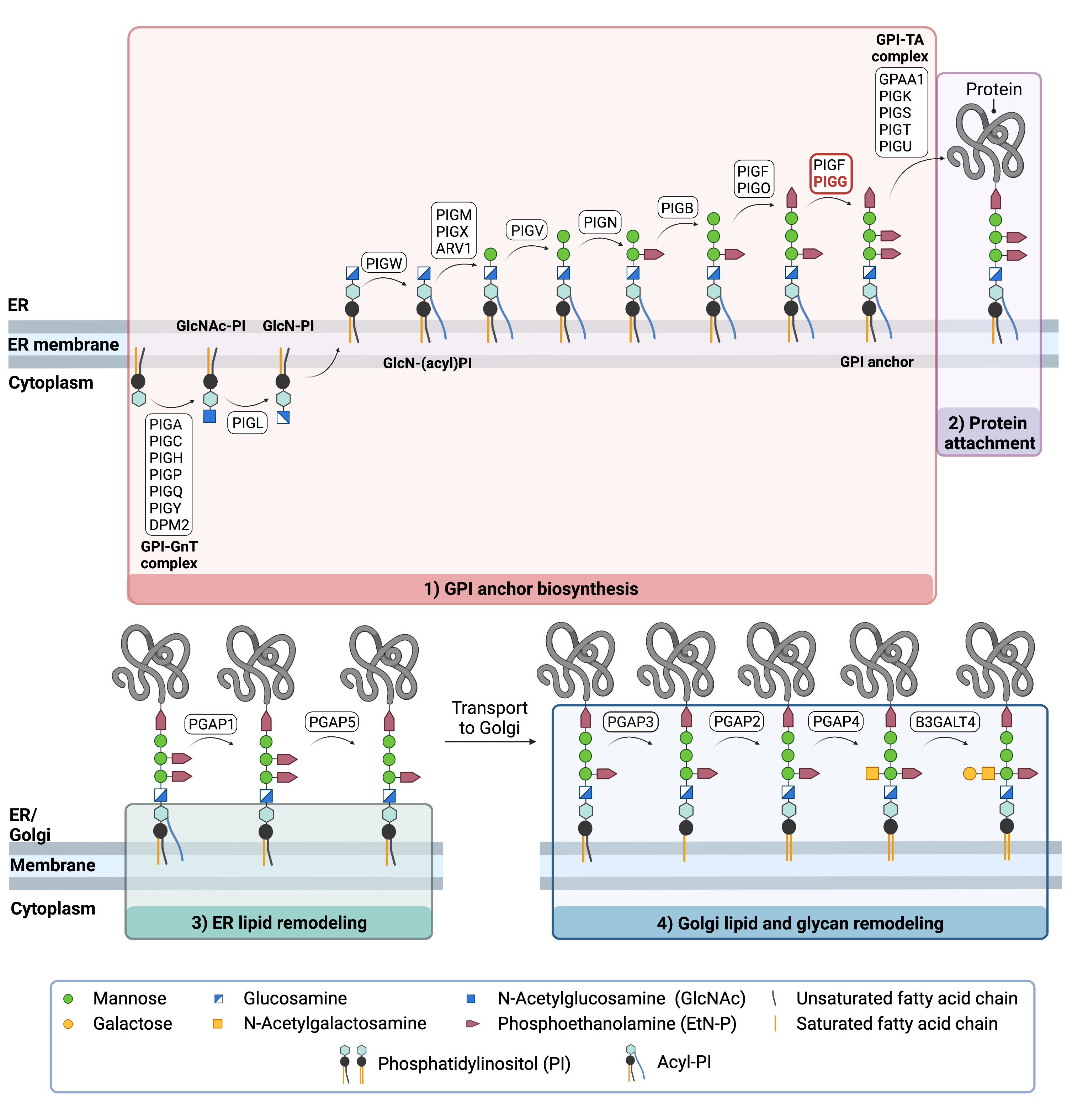
Figure 2. Overview of GPI-anchored protein biosynthesis.
GPI-anchored protein biosynthesis relies on a series of enzymatic reactions. First, the GPI anchor core is built on the ER membrane, where enzymes modify the lipid and glycan portions, generating PI-GlcN-Man3-EtN-P. The protein is then attached to the GPI anchor in the ER, before further modifications are made to the lipid and glycan portions in the ER and the Golgi. PIG-G forms a complex with PIG-F (GPI-EtNP transferase II) which catalyzes the attachment of the third EtN-P group to the GPI anchor in the ER .
GPI ANCHOR SYNTHESIS
The first stage of GPI anchor biosynthesis involves the stepwise construction of the GPI anchor. N-acetylglucosamine (GlcNAc) is added to the lipid phosphatidylinositol (PI), generating GlcNAc-PI. The transfer of GlcNAc is catalyzed by GPI GlcNAc transferase (GPI-GnT). Additional enzymes then further modify the GPI anchor structure by transferring three mannose residues and three EtN-P groups and PI-GlcN-Man3-EtN-P)2,4,8,10.PIG-G is stabilized by PIG-F, and it competes with PIG-O for binding to PIG-F13. The 5
PROTEIN ATTACHEMENT
Once the GPI anchor has been synthesized, it is transferred en bloc to a protein with a C-terminal GPI attachment signal sequence by GPI transamidase (GPI-TA). The five-protein enzyme complex catalyzes the simultaneous cleavage of the signal sequence and attachment of the GPI anchor to the newly synthesized protein7.
LIPID/GLYCAN REMODELING AND PROTEIN TRANSPORT
After the protein has been attached to the GPI anchor, both the glycan and lipid portion of the anchor undergo modifications (referred to as remodeling) in the ER and Golgi by post-GPI attachment to protein (PGAP) enzymes or the glycosyltransferase B3GALT4. The complete GPI-anchored protein is then transported to the plasma membrane where it associates with other GPI-anchored proteins in lipid rafts11.
Disease Mechanism
Mutations in the PIGG gene lead to loss of function of the enzyme, resulting in a failure to complete this GPI biosynthesis pathway and a reduction in functional GPI-anchored proteins on the cell surface4. GPI-anchored proteins have several critical functions in the cell such as adhesion molecules, receptors, and enzymes in signal transduction pathways. GPI-anchored proteins are known to be expressed during neurogenesis, and even a slight deficiency in GPI-anchored proteins results in defects in neuronal development2.
Mutations
The PIGG gene is located on the Chromosome 4 (4p16.3). Mutations that result in complete knockout or severely decreased activity phenotypically present with epileptic seizures, hypotonia, cerebellar atrophy, diminished deep tendon reflexes and dysmorphisms. Mutations that result in mildly decreased or slight to no decreased activity phenotypically present with epileptic seizures and autism spectrum disorder2.
Signs & Symptoms
Clinical Presentation
Individuals with PIGG-CDG typically develop signs and symptoms during infancy. PIGG-CDG is primarily characterized by developmental delay and intellectual disability and dysmorphic features. Symptoms of PIGG-CDG include1–3:
- Neurological– developmental delay, intellectual disability, low muscle tone (hypotonia), epileptic seizures (in the first two years of life), cerebellar atrophy, neurological manifestations, ataxia, nystagmus, strabismus, tremor autism spectrum disorder.
- Dysmorphic features– facial dysmorphism including long philtrum, shallow nose tip, crossed eyes (strabismus), short stature
A less common symptom of PIGG-CDG includes mitochondrial complex deficiency and diminished deep tendon reflexes.
Biochemical Abnormalities
Biochemical abnormalities have not been reported.
Classification
Diagnosis
GPI-related CDG should be considered in individuals presenting with early onset severe seizure disorders and dysmorphic facial features, even if transferrin and total N-glycan analysis are normal. As currently available screening tests for CDG will not reliably detect PIGG-CDG, diagnosis is typically achieved through genetic testing, either as part of an epilepsy panel or whole exome sequencing. PIGG-CDG may also be diagnosed by analyzing surface GPI-anchor proteins on blood cells and fibroblast cells by flow cytometry.
GPI-Anchored Protein Flow Cytometry
Individuals with PIGG-CDG may lack GPI-anchored proteins on the surface of their granulocytes, however patients have been reported with normal surface levels and structure of GPI-anchor proteins, even with near complete inactivation of PIG-G.
In contrast, testing patient derived fibroblast tissue have found that GPI-anchored proteins are decreased, with CD73 as the lowest marker. This suggests that Fibroblasts may be the best tissue for PIGG deficiency diagnosis2.
Biomarkers
There are no currently no known biomarkers specific to PIGG-CDG.
Prognosis
Prognosis of PIGG-CDG may vary depending on severity of an individual’s symptoms. The broad clinical spectrum may result in some patients with severe mutations presenting with intellectual disability and various neurological manifestations, while patients with less severe mutations presenting with autism spectrum disorder2.
Developmental delays, seizures, and uncoordinated movements (ataxia) symptoms seem to improve with age in most patients2. The oldest patient reported in the literature is 24 years old1.
Management
Management of symptoms may include combinations of physical therapy, occupational therapy, and palliative measures.
One patient reported with hemiplegic migraines that were controlled with amitriptyline. Another patient reported acute onset ataxic attacks with regression that was treated with high dose corticosteroids2.
Therapies
There are currently no treatment options available for PIGG-CDG.
However, pyridoxine, a form of vitamin B6, has been investigated as a treatment for GPI deficient patients with treatment-resistant epilepsy. Pyridoxine was investigated as a treatment for GPI disorders because it is thought that there may be a reduced level of intracellular pyridoxine, specifically in the brain, due to disrupted GPI-anchored protein function. For patients with PIGG-CDG, pyridoxine and/or pyridoxal phosphate have potential to be used for seizure treatment2.
Research Models
Various research models are available to study PIGG-CDG.
Yeast (S. cerevisae)
Yeast gene Gpi17 is homologous to human PIGG and is a member of the glycosyltransferase gene family. Deletion of Gpi17 in yeast has significant effects on cell growth and separation12.
Mouse (M. Musculus)
Pigg first allele knockout embryonic stem cells
Piggtm1a(EUCOMM)Wtsi and Piggtm2a(EUCOMM)Wtsi are first allele knockout embryonic stem cells. Phenotype data is not available (IMPC).
Chinese Hamster Ovary Cell Line
Coprecipitation of transfected CHO cells were used to demonstrate that human PIG-F interacts with PIG-G (GP17) and PIG-O. The interaction with PIG-F was determined to stabilize PIG-G and PIG-O during transferase reaction and PIG-G competes with PIG-O to bind with PIG-F13.
Human Cell Lines
PIGG-/- HEK293 cells
PIGG-/- HEK293 cells were generated using CRISPR/Cas9. These cells were used to confirm that a complete loss of PIG-G does not affect cell surface levels of GPI-anchor proteins1.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including PIGG-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Organizations
GPI-anchor CDG Community Facebook Group
Publications
PIGG-CDG Scientific Articles on PubMed
Additional Resources
Kinoshita Lab (GPI anchor pathway researchers)
References
- Makrythanasis, P. et al. Pathogenic Variants in PIGG Cause Intellectual Disability with Seizures and Hypotonia. American Journal of Human Genetics 98, 615–626 (2016).
- Tremblay-Laganière, C. et al. PIGG variant pathogenicity assessment reveals characteristic features within 19 families. Genetics in Medicine 23, 1873–1881 (2021).
- Lionel, A. C. et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genetics in Medicine 20, 435–443 (2017).
- Zhao, J. J. et al. Reduced cell surface levels of GPI-linked markers in a new case with PIGG loss of function. Human Mutation 38, 1394–1401 (2017).
- Wu, T. et al. The Glycosylphosphatidylinositol biosynthesis pathway in human diseases. Orphanet Journal of Rare Diseases 15, (2020).
- Kinoshita, T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biology 10, (2020).
- Kinoshita, T. Glycosylphosphatidylinositol (GPI) anchors: Biochemistry and cell biology: Introduction to a thematic review series. Journal of Lipid Research 57, (2016).
- Liu, Y. S. & Fujita, M. Mammalian GPI-anchor modifications and the enzymes involved. Biochemical Society Transactions 48, (2020).
- Englund, P. T. The Structure and Biosynthesis of Glycosyl Phosphatidylinositol Protein Anchors. Annual Review of Biochemistry62, (1993).
- Maeda, Y. & Kinoshita, T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Progress in Lipid Research 50, (2011).
- Fujita, M. & Kinoshita, T. Structural remodeling of GPI anchors during biosynthesis and after attachment to proteins. FEBS Letters 584, (2010).
- Benachour, A. et al. Deletion of GPI7, a yeast gene required for addition of a side chain to the glycosylphosphatidylinositol (GPI) core structure, affects GPI protein transport, remodeling, and cell wall integrity. Journal of Biological Chemistry 274, 15251–15261 (1999).
- Shishioh, N. et al. GPI7 is the second partner of PIG-F and involved in modification of glycosylphosphatidylinositol. J Biol Chem280, 9728–9734 (2005).