
Lay Summary
PIGN-CDG is a rare inherited condition that affects several systems in the body. PIGN-CDG is also commonly referred to as Multiple Congenital Anomalies-Hypotonia-Seizures syndrome type 1 (MCAHS1). There have been approximately 70 patients reported to date in the medical literature. PIGN-CDG is classified as a disorder of GPI anchor biosynthesis and is caused when an individual has mutations in both copies of their PIGN gene. The PIGN gene provides instructions for making a protein that participates in building GPI anchors, which are molecules that “anchor” certain proteins to the cell surface. The PIGN protein attaches a molecule called phosphoethanolamine to the GPI anchor which is important for its function. Mutations in the PIGN result in a reduced number of GPI-anchored proteins on the cell surface. Symptoms of PIGN-CDG being during pregnancy and infancy and are primarily characterized by delayed psychomotor development, low muscle tone, and seizures. Mutations in PIGN are also associated with Fryns syndrome, a multiple congenital anomalies syndrome that is characterized by defects in the diaphragm, distinctive facial features, underdevelopment of nail and finger bones and other associated anomalies. Survival beyond the neonatal period is rare for individuals with Fryns syndrome. PIGN-CDG is usually diagnosed through genetic testing, however testing for the presence of GPI-anchored proteins on blood cells can also help screen for PIGN-CDG. Seizure medication has been shown to be successful in some patient’s management of seizure symptoms. Pyridoxine is also being testing as a treatment, however current treatment is focused on the management of symptoms and preventing complications.
Overview
Phosphatidylinositol glycan class N congenital disorder of glycosylation (PIGN-CDG) is a rare autosomal recessive genetic disorder. The first reported case of PIGN-CDG was in 2011, and there are approximately 70 confirmed cases to date1–14. PIGN-CDG is also commonly referred to as Multiple Congenital Anomalies-Hypotonia-Seizures syndrome type 1 (MCAHS1). Mutations in PIGN are also associated with Fryns syndrome, which is a condition with multiple abnormalities and few patients surviving the neonatal period.
The PIGN gene encodes an enzyme that catalyzes the transfer of phosphoethanolamine (EtN-P) to the first mannose in GPI during GPI anchor protein biosynthesis15. GPI anchors are an important mechanism of attachment for proteins to the cell surface, and these proteins are known as GPI-anchored proteins. Mutations in PIGN result in reduced expression of GPI-anchored proteins, which are critical to the development and function of many organs, especially the nervous system7,15.
Symptoms may begin during pregnancy and during infancy and the characteristic presentation includes seizures, delayed psychomotor development, congenital anomalies of the cardiac, urinary tract and gastrointestinal system, and facial abnormalities8,12,14,16,17. Although a definitive diagnosis can only be achieved through genetic sequencing, analyzing blood cells for an absence of GPI-anchored proteins can help diagnose disorders of GPI anchor biosynthesis. Currently treatment is limited to management of seizure symptoms with seizure medication, as well as testing pyridoxine as a treatment option17.
Synonyms
- Congenital Disorder of Glycosylation due to PIGN deficiency
- Multiple Congenital Anomalies-hypotonia-seizures syndrome type 1 (MCAHS1)
- GPI biosynthesis defect type 3 (GPIBD3)
- Fryns syndrome
Inheritance
PIGN-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The PIGN gene encodes ethanolamine phosphate transferase (PIG-N), which is one of several enzymes involved in glycosylphosphatidylinositol (GPI) anchor biosynthesis. In the endoplasmic reticulum (ER), PIG-N catalyzes the addition of phosphoethanolamine (EtN-P) to the first mannose in GPI.
GPI-ANCHORED PROTEIN BIOSYNTHESIS
GPI-anchored protein biosynthesis is one of the major glycosylation pathways that attach glycans to lipid molecules within cells. Many proteins, called GPI-anchored proteins, are attached to the cell surface by GPI anchors.
The core structure of GPI consists of phosphatidylinositol (PI), glucosamine (GlcN), three mannose sugars (Man)3, and three phosphoethanolamine (EtN-P) connected to each other in that sequence (Figure 1). GPI-anchored proteins are attached to the GPI by forming a bond between the EtN-P group of the GPI core and the C-terminus of newly synthesized proteins in the ER lumen15.
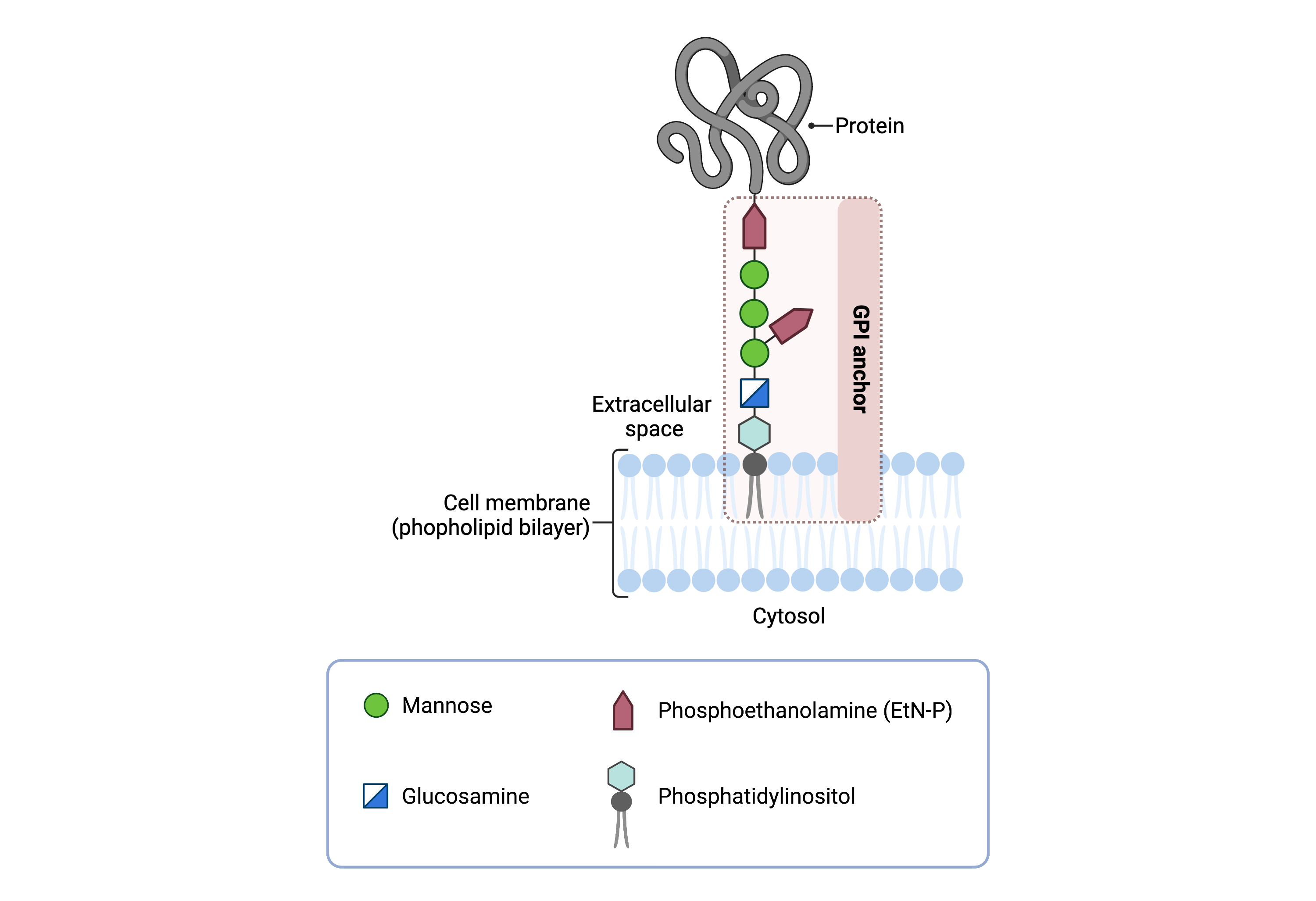
Figure 1. Overview of GPI-anchored protein structure.
The GPI anchor is added to proteins, generating GPI-anchored proteins. The GPI anchor section lodges into the membrane, attaching the protein to the membrane. The GPI anchor involves phosphatidylinositol, glucosamine, mannose and phosphoethanolamine.
The generation of a GPI-anchored proteins is a multi-step process involving more than 30 enzymes and can be divided into the following steps (Figure 2) 18–20:
- GPI anchor synthesis
- Protein attachment
- Lipid/glycan remodelling and protein transport
GPI anchor synthesis and protein attachment is largely carried out by a series of enzymes encoded by the PIG genes, while enzymes encoded by the PGAP genes facilitate remodelling of the GPI anchored protein.
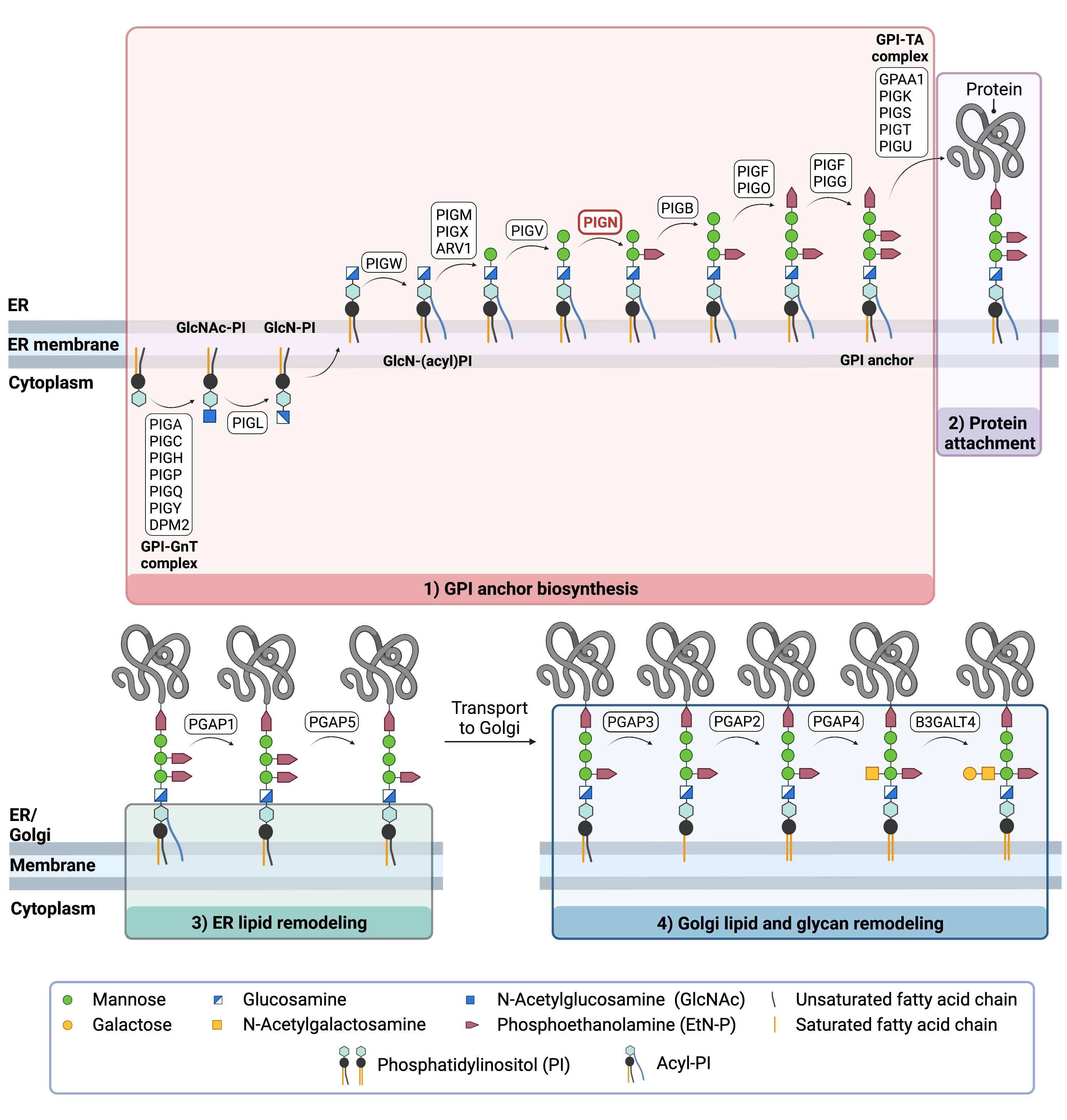
Figure 2. Overview of GPI-anchored protein biosynthesis and role of PIG-N.
GPI-anchored protein biosynthesis involves a series of enzymatic reactions. First, the GPI anchor core is built on the ER membrane, where enzymes modify the lipid and glycan portions, generating PI-GlcN-Man3-EtN-P. The protein is then attached to the GPI anchor in the ER, before further modifications are made to the lipid and glycan portions in the ER and the Golgi. PIG-N catalyzes the transfer of ethanolamine phosphate to the first mannose residue of the GPI anchor in ER lumen.
GPI Anchor Synthesis
The first stage of GPI-anchored protein biosynthesis involves the stepwise construction of the GPI anchor, which begins on the cytoplasmic side of the ER. The first step involves the transfer of the monosaccharide N-acetylglucosamine (GlcNAc) from nucleotide sugar UDP-GlcNAc to the lipid phosphatidylinositol (PI), generating GlcNAc-PI. The transfer of GlcNAc is catalyzed by GPI GlcNAc transferase (GPI-GnT), a 7-subunit protein complex15.
GlcNAc-PI then has its acetyl (Ac) group removed from the GlcNAc sugar, generating glucosamine phosphatidyl inositol (GlcN-PI) which is transported into the luminal side of the ER19,21. Within the ER, a fatty acid chain is attached to GlcN-PI, generating GlcN-(acyl)PI. The final steps of the GPI anchor synthesis involve the sequential addition of 3-4 mannose sugars and three EtN-P groups by several PIG enzymes. PIG-N catalyzes the transfer of the EtN-P from diradyl phosphatidylethanolamines (PE) onto the first mannose residue of GPI15. PIG-B and PIG-F (which forms catalytic complexes with both PIG-O and PIG-G), catalyze the attachment of two additional EtN-P groups onto GPI. The completed GPI anchor core, PI-GlcN-Man3-EtN-P, is then ready to be attached to a protein19,22.
Protein Attachment
Once the GPI anchor has been synthesized, it is transferred en bloc to a protein with a C-terminal GPI attachment signal sequence by GPI transamidase (GPI-TA). The five-protein enzyme complex catalyzes the simultaneous cleavage of the signal sequence and attachment of the GPI anchor to the newly synthesized protein23,24 .
LIPID/GLYCAN REMODELLING AND PROTEIN TRANSPORT
After the protein has been attached to the GPI anchor, both the glycan and lipid portion of the anchor undergo modifications (referred to as remodelling) in the ER and Golgi by post-GPI attachment to protein (PGAP) enzymes. In the ER, remodelling includes the removal of the fatty acid chain and one EtN-P group, which enable the protein to be transported to the Golgi. In the Golgi, the lipid portion of the anchor is exchanged with other lipids that better enable the protein to associate with lipid rafts in the plasma membrane. Some GPI-anchored proteins undergo further modification by the addition of an N-acetlygalactosamine (GalNAc) glycan before being transported to the cell membrane. The complete GPI-anchored protein is then transported to the plasma membrane where it associates with other GPI-anchored proteins in lipid rafts25.
Disease Mechanism
Mutations in the PIGN gene lead to a reduction in functional GPI-anchored proteins and reduced expression pf CD59 on the fibroblast cell surface7. GPI anchor proteins have several critical functions in the cell such as embryogenesis, immune response, and neurogenesis15.
Mutations
The PIGN gene is located on chromosome 18 (18q21.33). Almost 70 pathogenic variants in the PIGN gene have been reported to date11. Almost all PIGN variants causing MCAHS1 are biallelic missense or loss of function10. Recurrent mutation c.932T>G p.(L311W) is associated with generalized seizures11. Individuals with biallelic truncated PIGN variants present with a severe clinical phenotype which includes early onset of intractable seizures and death in utero or shortly after birth26,27. Patients with non-truncating variants appear to have a milder phenotype with fewer dysmorphic features, as these mutations maintain some residual protein function26. Mutations to PIGN are also associated with Fryns Syndrome, an autosomal recessive condition characterized by congenital diaphragmatic hernia and other congenital anomalies9,10,28.
Signs & Symptoms
Clinical Presentation
Individuals with PIGN-CDG typically develop signs and symptoms during pregnancy and throughout infancy. PIGN is expressed widely in different tissues, resulting in diverse clinical phenotypes8,16,17. PIGN-CDG is primarily characterized by neonatal hypotonia, seizures, delayed or lack of psychomotor development, and multiple congenital anomalies. Symptoms of PIGN-CDG include8,12,14,17,29:
- Neurological symptoms – developmental delay, intellectual disability, low muscle tone (hypotonia), weak muscles (hyporeflexia), overactive reflexes (hyperreflexia), epileptic seizures, cerebellar and cerebral atrophy.
- Congenital anomalies– gastrointestinal system (gastroesophageal reflux), cardiac system (atrial septal defect, persistent foramen ovale), urinary tract, diaphragmatic defects
- Facial abnormalities – cleft palate, thin lips, small nose, depressed nasal bridge, epicanthal folds, large fleshy ears, low set ears, long philtrum, small lower jaw (micrognathia)
Less common symptoms of PIGN-CDG includes visual impairment and involuntary side-to-side movement of eyes (nystagmus), underdeveloped kidney, diaphragmatic hernia, underdeveloped lungs. Fryns Syndrome is characterized by congenital diaphragmatic hernia, abnormal development of fingers, dysmorphic facial features, and other malformations30.
Biochemical Presentation
One patient has been reported with elevated serum alkaline phosphatase17.
Classification
PIGN-CDG is classified as a disorder of GPI anchor biosynthesis.
Diagnosis
PIGN-CDG should be considered in individuals presenting with developmental disability, hypotonia, congenital diaphragmatic hernia, epilepsy and other congenital anomalies, even if transferrin and total N-glycan analysis are normal7. As currently available screening tests for CDG will not reliably detect PIGN-CDG, diagnosis is typically achieved through genetic testing, either as part of an epilepsy panel or whole exome sequencing. PIGN-CDG may also be primarily screened for by analyzing surface GPI-anchor proteins on blood cells by flow cytometry.
GPI-Anchored Flow Cytometery
Individuals with PIGN-CDG lack GPI-anchor proteins on the surface of their granulocytes, a type of white blood cell. Analysis of FLAER and anti-CD59 by flow cytometry has been demonstrated to aid in confirming GPI anchor biosynthesis defects, consistent with PIGN-related disease26.
Biomarkers
There are currently no known biomarkers specific to PIGN-CDG.
Prognosis
Prognosis of PIGN-CDG may vary depending on severity of an individual’s symptoms. Onset of epilepsy typically occurs before 1 year of age can be therapy resistant. Cerebellar atrophy is typically progressive. Early death has been reported in a number of patients ranging from infancy to adulthood. For individuals with Fryns syndrome, survival beyond the neonatal period is rare and those who survive past this stage have documented developmental delays 2,7,11,14.
Management
Management of symptoms may include combinations of physical therapy, occupational therapy, vison and speech therapy and palliative measures. Gastronomy therapy may be used to overcome feeding problems. In some patients, seizures are treatable with anti-epileptic medication. Surgical intervention is required to manage diaphragmatic hernias.
Therapies
Levetiracetam and other antiepileptic drugs have been used to control patient seizures with some success. Pyridoxine, a form of vitamin B6, has also been tested in conjunction with seizure medication as a treatment for PIGN-CDG, however, the efficacy of pyridoxine is difficult to assess at this time17.
Research Models
Research models are available to study PIGN-CDG include yeast, worm, mouse models, and human cell lines.
Yeast (S. cerevisiae)
Yeast MCD4 (PIGN ortholog) mutants show morphological defects, defective bud emergence, polarized growth, and defective transportation from the ER to the Golgi. The lumenal domain of Mcd4 contains three conserved motifs that are found in mammalian phosphodiesterases31.
Worm (C. elegans)
C. elegans pign-1 is an ortholog of human PIG-N. It is involved in GPI-anchor biosynthesis and protein quality control in the ER. Pign-1 mutants an accumulation of secretory protein aggregates in the ER, demonstrating that pign-1 is required for quality control of proteins in the ER. Mutations to pign-1 that correspond to mutations in MCAHS1 patients are also shown to cause protein aggregation in the ER, which suggests the non-canonical function of PIG-N may play a role in the disease mechanism of MCAHS1 32.
Pign-1 mutant, xyz11, was used to investigate glycosylation site N127. It was found that this glycosylation site is important for the non-canonical function of Pig-n, preventing protein aggregation in cells33.
Mouse (M. musculus)
Pignem1(IMPC)Mbp exon deletion
Heterozygous mice with exon deletion live to early adulthood and show abnormal skin morphology, as well as enlarged spleen, small kidney, and small liver (MGI).
Pignm1Nisw/J mouse embryonic fibroblasts (MEF)
Mutant mice show a holoprosencephaly like phenotype, including anterior truncations in mutant embryos. This m1Nisw/J (also called Gnz) mutation disrupts the Pign gene and results in mislocalization of GPI anchor proteins34.
Human Cell lines
PIGN -/- HEK293T knockout cells
PIGN-knockout cells (PIGN-/-) were generated from HEK293 cells using the CRISPR/Cas System which displayed a reduced expression of GPI-anchored protein CD593.
The PIGN-knockout HEK293 cells generated by Ohba et al., 2014 were used to investigate the role of PIGN in protein quality control. Similar to the observations in C. elegans pign-1 mutants, protein aggregation was observed in human cells with deficient PIGN32.
Patient-Derived Fibroblast lines
The expression of GPI-linked protein CD59 on fibroblasts from patients as compared to that in a control individual showed a 10-fold reduction in expression7.
Clinical Studies
Acitve
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including PIGN-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Organizations
GPI-anchor CDG Community Facebook Group
PIGN GPI-biosynthesis defects Facebook Group
Publications
PIGN-CDG & Fryns syndrome Scientific Articles on PubMed
Additional Resources
PIGN-CDG Infographic
Kinoshita Lab (GPI anchor pathway researchers)
IEMbase
OMIM
Orphanet
Genetic Testing Registry
ClinVar
NIH
GeneCards
UniProt
References
- Nakagawa, T. et al. A novel PIGN mutation and prenatal diagnosis of inherited glycosylphosphatidylinositol deficiency. American Journal of Medical Genetics Part A 170, 183–188 (2016).
- Fleming, L. et al. Genotype-phenotype correlation of congenital anomalies in multiple congenital anomalies hypotonia seizures syndrome (MCAHS1)/ PIGN -related epilepsy. American Journal of Medical Genetics Part A 170, 77–86 (2016).
- Ohba, C. et al. PIGN mutations cause congenital anomalies, developmental delay, hypotonia, epilepsy, and progressive cerebellar atrophy. Neurogenetics 85–92 (2014)
- Pagnamenta, A. T. et al. Analysis of exome data for 4293 trios suggests GPI-anchor biogenesis defects are a rare cause of developmental disorders. European Journal of Human Genetics 25, 669–679 (2017).
- Jezela-Stanek, A. et al. Congenital disorder of glycosylphosphatidylinositol (GPI)-anchor biosynthesis—The phenotype of two patients with novel mutations in the PIGN and PGAP2 genes. European Journal of Paediatric Neurology 20, 462–473 (2016).
- Brady, P. D. et al. Exome sequencing identifies a recessive PIGN splice site mutation as a cause of syndromic Congenital Diaphragmatic Hernia. European Journal of Medical Genetics 57, 487–493 (2014).
- Maydan, G. et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. Journal of Medical Genetics 48, 383–389 (2011).
- Couser, N. L. et al. The Phenotype of Multiple Congenital Anomalies-Hypotonia-Seizures Syndrome 1: Report and Review. Am J Med Genet A 167A, 2176 (2015).
- McInerney-Leo, A. M. et al. Fryns Syndrome Associated with Recessive Mutations in PIGN in two Separate Families. Human Mutation 37, 695–702 (2016).
- Alessandri, J.-L. et al. Recessive loss of function PIGN alleles, including an intragenic deletion with founder effect in La Réunion Island, in patients with Fryns syndrome. European Journal of Human Genetics 26, 340–349 (2018).
- Bayat, A. et al. PIGN encephalopathy: Characterizing the epileptology. Aleksandra Jezela-Stanek 25, (2022).
- Thiffault, I. et al. Hypotonia and intellectual disability without dysmorphic features in a patient with PIGN-related disease. BMC Medical Genetics 18, 1–5 (2017).
- Xiao, S. Q. et al. Case Report: Compound Heterozygous Phosphatidylinositol-Glycan Biosynthesis Class N ( PIGN) Mutations in a Chinese Fetus With Hypotonia-Seizures Syndrome 1. Front Genet 11, (2020).
- Khayat, M. et al. A PIGN Mutation Responsible for Multiple Congenital Anomalies-Hypotonia-Seizures Syndrome 1 (MCAHS1) in an Israeli-Arab Family. American Journal of Medical Genetics Part A 176–182 (2017).
- Kinoshita, T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biology 10, (2020).
- Knaus, A. et al. Characterization of glycosylphosphatidylinositol biosynthesis defects by clinical features, flow cytometry, and automated image analysis. Genom2 Med. 17, (2018).
- Jiao, X. et al. Analyzing clinical and genetic characteristics of a cohort with multiple congenital anomalies-hypotonia-seizures syndrome (MCAHS). Orphanet J Rare Dis 15, (2020).
- Kinoshita, T. Glycosylphosphatidylinositol (GPI) anchors: Biochemistry and cell biology: Introduction to a thematic review series. Journal of Lipid Research 57 (1), 4-5 (2016).
- Liu, Y. S. & Fujita, M. Mammalian GPI-anchor modifications and the enzymes involved. Biochemical Society Transactions. 48 (3), 1129-1138. (2020).
- Englund, P. T. The Structure and Biosynthesis of Glycosyl Phosphatidylinositol Protein Anchors. Annual Review of Biochemistry 62, (1993).
- Bayat, A. et al. Deciphering the premature mortality in PIGA-CDG – An untold story. Epilepsy Research 170, (2021).
- Maeda, Y. & Kinoshita, T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Progress in Lipid Research 50 (4):411-24 (2011).
- Ohishi, K., Nagamune, K., Maeda, Y. & Kinoshita, T. Two Subunits of Glycosylphosphatidylinositol Transamidase, GPI8 and PIG-T, Form a Functionally Important Intermolecular Disulfide Bridge. Journal of Biological Chemistry 278, 13959–13967 (2003).
- Ohishi, K. PIG-S and PIG-T, essential for GPI anchor attachment to proteins, form a complex with GAA1 and GPI8. The EMBO Journal 20, 4088–4098 (2001).
- Fujita, M. & Kinoshita, T. Structural remodeling of GPI anchors during biosynthesis and after attachment to proteins. FEBS Letters 584(9): 1670-7 (2010).
- Thiffault, I. et al. Hypotonia and intellectual disability without dysmorphic features in a patient with PIGN-related disease. BMC Med Genet 8(1):124 (2017).
- Misra, H. S. et al. Case Report: Compound Heterozygous Phosphatidylinositol-Glycan Biosynthesis Class N (PIGN) Mutations in a Chinese Fetus With Hypotonia-Seizures Syndrome 1. Front Genet 11:594078. (2020)
- Thompson, M. D. & Cole, D. E. Recessive PIGN Mutations in Fryns Syndrome: Evidence for Genetic Heterogeneity. Human Mutation 37, 621–621 (2016).
- OMIM Entry - # 614080 - MULTIPLE CONGENITAL ANOMALIES-HYPOTONIA-SEIZURES SYNDROME 1; MCAHS1. https://www.omim.org/entry/614080.
- McInerney-Leo, A. M. et al. Fryns Syndrome Associated with Recessive Mutations in PIGN in two Separate Families. Human Mutation 37, 695–702 (2016).
- Gaynor, E. C. et al. MCD4 Encodes a Conserved Endoplasmic Reticulum Membrane Protein Essential for Glycosylphosphatidylino-sitol Anchor Synthesis in Yeast. Molecular Biology of the Cell 10 (1999).
- Ihara, S. et al. PIGN prevents protein aggregation in the endoplasmic reticulum independently of its function in the GPI synthesis. J Cell Sci 130(3):602-613 (2017).
- Narimatsu, T. & Ihara, S. New allele of C. elegans gene pign-1, named as xyz11. microPublication Biology 2019, (2019).
- McKean, D. M. & Niswander, L. Defects in GPI biosynthesis perturb Cripto signaling during forebrain development in two new mouse models of holoprosencephaly. Biology Open 1, 874–883 (2012).