
Lay Summary
SLC39A8-CDG, formerly known as CDG-Iln, is a rare inherited condition that affects many parts of the body. To date, 17 cases have been reported in the medical literature. SLC39A8-CDG is classified as disorder of multiple glycosylation pathways. SLC39A8-CDG is caused when an individual has mutations in both copies of their SLC39A8 gene which provides instructions for making a type of protein called a solute carrier (SLC) that helps transport the mineral manganese into the cell. Manganese is an essential nutrient that plays an important role in many biological processes, including glycosylation. Specifically, enzymes that attach the simple sugar galactose onto sugar chains on proteins require manganese to function properly. Mutations in the SLC39A8 gene result in manganese deficiency and impaired glycosylation as the protein is unable to transport manganese into the cell. Symptoms of SLC39A8-CDG begin at infancy and are primarily characterized by global developmental delay, low muscle tone, poor postural control, gastrointestinal and feeding problems, and dysmorphic features. Some patients with SLC39A8-CDG may present with features similar to Leigh syndrome, a progressive neurodegenerative disorder that is caused by defects in the mitochondria. Screening tests are available for SLC39A8-CDG, but a definitive diagnosis is achieved through genetic testing. Currently, treatment via oral supplementation of galactose and/or manganese have been shown to improve symptoms of SLC39A8-CDG.
Overview
Solute Carrier Family 39 Member 8 congenital disorder of glycosylation (SLC39A8-CDG) is a rare autosomal recessive genetic disorder. The first reported case of SLC39A8-CDG was in 20151,2 and 17 confirmed cases have been reported to date1–5. The SLC39A8 gene encodes the manganese transporter protein, ZIP8, which plays a major role in regulating manganese homeostasis in the blood and tissues6,7. ZIP8 can be found on the plasma and organelle membranes, such as the Golgi and mitochondria, where it moves manganese across the membrane to the inside of a cell or organelle—ZIP8 can also transport other metal ions too, such as zinc and iron6. Manganese is an essential nutrient that plays a major biological role as a cofactor for a variety of enzymes, including galactosyltransferases, which attach galactose to glycoproteins or glycolipids in the Golgi during glycosylation, and mitochondrial enzymes1. Consequently, mutations in SLC39A8 can prevent sufficient levels of manganese getting inside the cell and organelles. These deficient manganese levels reduce the activity of manganese-dependent enzymes in the Golgi and mitochondria, including galactosyltransferases, which can lead to hypoglycosylation of proteins and cause SLC39A8-CDG6.
Symptoms of SLC39A8-CDG begin at infancy and the characteristic clinical presentation includes neurological defects, severe psychomotor disability, seizures, gastrointestinal and feeding problems, and dysmorphic features5. With SLC39A8 also located on the mitochondrial membrane and important for components of the mitochondria to function properly, some patients may present similar features to Leigh syndrome—a progressive neurodegenerative disorder that arises from defects in mitochondrial function8. A diagnosis may be determined through transferrin analysis, but a definitive diagnosis can only be achieved through genetic testing. Oral supplementation with galactose or manganese has been shown to improve biochemical and clinical features of SLC39A8-CDG1,3,9.
Inheritance
SLC39A8-CDG is an autosomal recessive disorder, where an affected individual inherits a mutation from each parent (asymptomatic carriers).
Synonyms
- CDG-Iln
- SLC39A8 deficiency
- Carbohydrate deficient glycoprotein syndrome type Iln
- Congenital disorder of glycosylation type 2n
Gene Function
ZIP8 (zinc and iron-related protein 8) is found at the plasma membrane and organelle membranes, such as the mitochondria, lysosome, Golgi, and endoplasmic reticulum (ER). ZIP8 helps maintain manganese homeostasis by permitting manganese entry into the cell and different intracellular organelles, while also regulating manganese levels in the blood or surrounding tissue10.
Manganese is a cofactor for various glycosylation and mitochondrial enzymes, such as β-1,4-galactosyltransferase (B4GALT1) and mannosylglycoprotein N-acetyl-glucosaminyltransferase 3 (MGAT3), which are involved in glycosylation in the Golgi, and manganese superoxide dismutase (MnSOD) which is involved in mitochondrial function. ZIP8 helps transport manganese into the Golgi and mitochondria for these enzymes to function5,7.
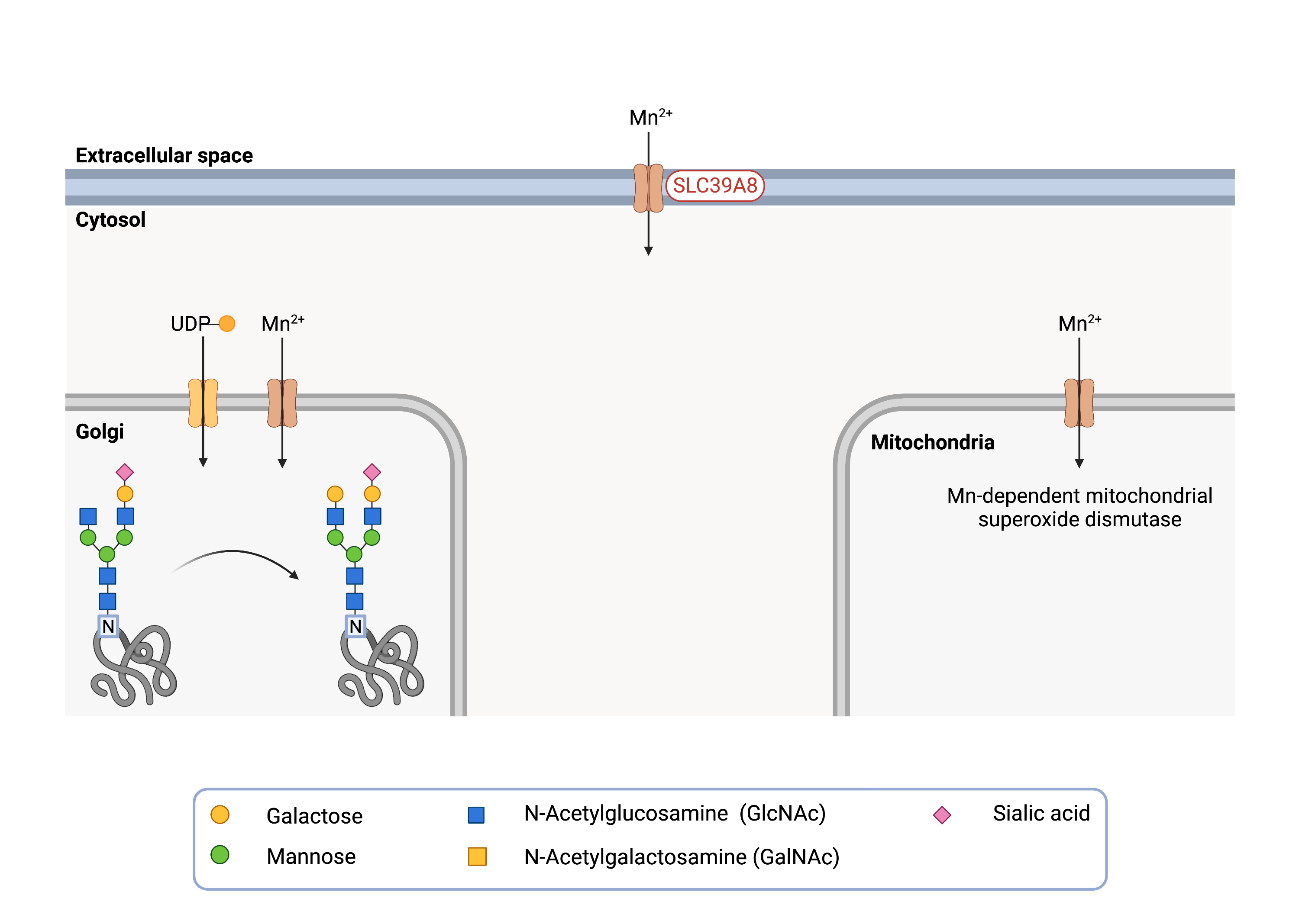
Figure 1. Role ofSLC39A8 in glycosylation.
SLC39A8 is a manganese transporter that moves manganese across the cell membrane and other cell compartments, such as the Golgi and mitochondria. Manganese ions are needed for different glycosylation components, such as the β-1,4-galactosyltransferase, and for mitochondria functioning, such as the Mn-dependent mitochondrial superoxide dismutase.
Glycosylation in the Golgi
A variety of different protein and lipid glycosylation pathways involve glycosylation and modification steps in the Golgi, such as N-glycosylation, where an oligosaccharide is attached to the asparagine (Asn; N) amino acid residue of a protein. N-glycosylation begins in the ER, where an oligosaccharide is synthesised on the lipid carrier dolichol and then attached to the protein11. Following attachment of the oligosaccharide to the protein, the N-glycoprotein is transported to the Golgi where the N-glycan undergoes branching through attachment of additional monosaccharides. These monosaccharides are attached to the glycan chain by glycosyltransferases. Some of these glycosyltransferases need to bind to manganese to be active, requiring ZIP8 to transport manganese into the Golgi. For example, B4GALT1 catalyzes the transfer of the galactose from UDP-galactose to N-glycans in the Golgi1,12. To be active, the B4GALT1 enzyme has two sites that must bind to manganese. In addition, a separate enzyme, MGAT3, requires manganese to function properly. MGAT3 catalyzes the transfer of N-acetylglucosamine (GlcNAc) from UDP-GlcNAc to a mannose residue on the N-glycan chain4,13.
Mitochondrial Function
Manganese is also a cofactor for MnSOD, a mitochondrial reactive oxygen species scavenger. Reactive oxygen species (ROS) are highly reactive chemicals that can cause damage to DNA, proteins, and lipids12. ROS can impair mitochondrial function by damaging mitochondrial DNA that generates proteins needed for the respiratory chain and energy production and mitochondrial enzymes. To scavenge ROS, MnSOD is dependent on manganese—relying on ion transporters like ZIP8 to move manganese into the cell and mitochondria5.
Disease Mechanism
SLC39A8-CDG is caused by manganese deficiency and hypoglycosylation. Mutations in the SLC39A8 gene render ZIP8 unable to localize to the plasma membrane and transport manganese into the cell, resulting in an intracellular manganese deficiency1,6. Galactosylation enzymes involved in N-glycosylation, such as B4GALT1 and MGAT3, are activated by manganese and have no/reduced enzyme activity in the absence of intracellular manganese, resulting in abnormal or hypoglycosylation of proteins1. Mutations in SLC39A8 have also been shown to reduce levels of mitochondrial manganese, which leads to impaired activity of manganese-dependent mitochondrial superoxide dismutase MnSOD, leading to an increase in oxidative stress6. Mutations of SLC39A8 reduce levels of mitochondrial manganese and MnSOD activity, along with increased ROS levels6. Additionally, SLC39A8-CDG can present similar clinical features to Leigh syndrome, which is a disorder associated with defects in mitochondrial energy production. Some pathogenic variants of SLC39A8 have been found to affect mitochondrial enzyme activity and energy production, reducing pyruvate dehydrogenase and respiratory chain complex (IV; II and III) activity6.
Mutations
The SLC39A8 gene is located on chromosome 4 (4p24) and several mutations in SLC39A8 that give rise to SLC39A8-CDG have been identified. The most common variant rs13107325 (C > T; A391T) is associated with an increased risk of other neurological disorders, such as drug-resistant epilepsy, intellectual disability5, and in some reports, schizophrenia14.
Mutations in SLC39A8 are also associated with Leigh-like syndrome clinical features, such as the pathogenic SLC39A8 variant: homozygous c.338G>C; p.(Cys113Ser)5,8.
Signs & Symptoms
Clinical Presentation
Individuals with SLC39A8-CDG typically develop signs and symptoms during infancy. SLC39A8-CDG is primarily characterized by a psychomotor disability, severe seizures, cerebellar atrophy, skull abnormalities. Affected individuals may also display clinical features similar to Leigh syndrome. The characteristic clinical presentations of SLC39A8-CDG include1–3,5:
- Neurological – global developmental delay, intellectual disability, low muscle tone (hypotonia), difficulty with balance (ataxia), poor postural control (dystonia), dyskinetic movements of the oral region, cerebral/cerebellar atrophy, and epilepsy/seizures
- Gastrointestinal – feeding difficulties and intestinal problems may lead to a failure to gain weight and slower than normal growth (failure to thrive)
- Ophthalmological – misaligned or crossed eyes (strabismus)
- Dysmorphic features – broad forehead, excess hair around the mouth and chin, upturned nose, thin lips, smooth philtrum, skeletal abnormalities, and scoliosis
Individuals with SLC39A8-CDG may present with features of Leigh syndrome-like mitochondrial disease which include progressive neurodegeneration (basal ganglia, thalami, white matter), intellectual and motor developmental delay, and mitochondrial respiratory chain or pyruvate dehydrogenase complex deficiency3.
Biochemical Abnormalities
Biochemical abnormalities observed in individuals with SLC39A8-CDG include severely reduced or undetectable levels of manganese in blood and increased manganese levels in urine1–3,5.
Classification
SLC39A8-CDG is classified as a disorder of multiple glycosylation pathways and within this group a disorder of Golgi homeostasis.
Under the former CDG classification system, SLC39A8-CDG is classified as a Type 2 CDG, which arise due to defects in the processing of N-glycans attached to proteins.
Diagnosis
SLC39A8-CDG may be suspected when patients present with developmental delay, severe intellectual disability, cerebral/cerebellar atrophy, muscular hypotonia, seizures, low/undetectable manganese levels in blood, and increased manganese levels in urine4,5. SLC39A8-CDG should also be suspected in patients presenting with Leigh syndrome symptoms that does not have a genetic confirmation. Screening in suspected patients typically begins with a blood test to analyze serum transferrin. Profiling of total serum N-glycans may also be performed and can aid in confirming diagnosis. However, diagnosis confirmation typically requires genetic testing of affected individuals5.
Transferrin Analysis
Most affected individuals will show a type 2 pattern by transferrin isoelectric focusing (TIEF) or capillary electrophoresis analysis of transferrin. Type 2 patterns have increased monosialo-, disialo- and trisialo-transferrin5. Transferrin analysis also show a decrease in galactose residues1,5. However, as some patients showed normal transferrin patterns, transferrin analysis alone is not reliable to diagnose SLC39A8-CDG. Mass spectrometry (MALDI-TOF) has been shown to detect age-related changes in transferrin glycosylation in some patients4,5.
Total Serum N-Glycan Analysis
A distinct N-glycome profile was detected by MALDI-TOF in patients who had normal or slightly abnormal transferrin patterns, showing reduced levels of bisected N-glycans and elevated levels of asialo-agalactosylated N-glycan (A2G1S1)4,5.
Biomarkers
There are no currently no known biomarkers specific to SLC39A8-CDG.
Prognosis
It is difficult to determine the long-term prognosis of SLC39A8-CDG as most patients reported in the medical literature are young. Prognosis of SLC39A8-CDG may vary depending on the severity of an individual’s symptoms. Severe cases include global developmental delay with severe intellectual disability, seizures, cerebellar atrophy, and recurrent infections5. As of 2015, the oldest patient recorded in medical literature is 23 years old2.
Management
Management of symptoms may include combinations of physical therapy, occupational therapy, speech or vision therapy, and palliative measures.
Therapies
Supplements such as galactose or manganese-II-sulfate have been found to improve biochemical (serum transferrin and manganese levels in blood and urine) as well as clinical features (motor abilities, muscle strength, ataxia, postural control, and epilepsy). Galactose supplements aim to correct hypogalactosylated proteins, by supplying increased levels of galactose for B4GALT1. Manganese-II-sulfate increase intracellular levels of manganese to restore manganese-dependent functions in the cell – such as mitochondrial enzymes – that cause other clinical abnormalities1,3,5,9.
Galactose Supplementation
Oral galactose supplementation (alone or in combination with uridine) has been shown to improve galactosylation and normalise glycosylation, with improvements seen 14 days after initiating treatment1,3,16.
Manganese-II-sulfate Supplementation
Successful treatment with dietary manganese-II-sulfate (MnSO4) supplementation has been reported in two patients. When treated with 15–20 mg of MnSO4 per kg of bodyweight, enzyme dysfunction appeared to be completely resolved as well as improvement of some symptoms, specifically motor abilities, hearing, vision, swallowing, and neurological symptoms such as seizures. MnSO4 was also found to improve biochemical features, restoring manganese levels in blood and urine and improving transferrin glycosylation9.
Research Models
Several research models have been generated including mouse models and human cell lines.
Mouse (M. musculus)
Homozygous Slc39a8-/-constitutive knockout mouse
Mutant mice show pre-weaning lethality (complete penetrance) (IMPC) and display impaired cardiovascular function, absence of sternum, small chest cavity, and a small liver17.
Slc39a8-inducible global knockout mouse
Slc39a8-inducible global knockout (ZIP8-iKO) mice show decreased manganese levels in multiple organs and whole blood sample18. Additionally, mutant mice show defective protein N-glycosylation. ZIP8-iKO mice were used with ZIP8-LSKO mice to compare a global knockout and liver-specific knockout of ZIP8. The study determined that hepatic ZIP8 regulates whole-body manganese homeostasis18.
Slc39a8 liver-specific knockout mouse
Slc39a8-liver-specific-knockout mice (ZIP8-LSKO), similar to ZIP8-iKO, show decreased manganese levels in multiple organs and whole blood samples. Mutant mice show defective protein N-glycosylation18.
Hypomorphic Slc39a8 knockout mouse
Reduced mRNA and protein levels of ZIP Zn2+/(HCO3-)2 symporter is observed in several tissues of the neonate mutants as well as reduced zinc and iron levels and embryonic and neonatal lethality. Surviving offspring were pale, presented growth arrest, severe anemia, hypoplastic spleen, and hypoplasia of liver, kidney, lung, and lower extremities. This experimental model is useful for studying ZIP8 function in the placenta, yolk sac, and fetal tissues7.
Slc39a8 lung-specific knockout mouse
BTZIP8-3 mice exhibit an increase in ZIP8 mRNA and protein expression in every tissue type examined, with the lung having the highest ZIP8 expression. It was discovered that BTZIP-3 lung has clustered filament actin (F-actin), which is not observed in wild-type mice19. Additionally, upregulated expression of proliferation biomarker Ki67 was seen in BTZIP8-3 lungs, suggesting a role of ZIP8 in regulating F-actin and promoting proliferation. Transcription factor NF-kB p65 was also shown to be elevated in BTZIP8-3 lung via western blotting. Signalling of molecular Snail2 (a transcription factor that regulates expression of E- and N-cadherin) was also shown to be regulated by ZIP8 in BTZIP8-3 mice; Snail2 expression was significantly increased, as well as Snail2 mRNA, in the lung19.
Mouse embryonic fibroblast (MEF) cells
Over expression of ZIP8 in mouse embryonic fibroblasts (MEFs) resulted in morphological change and re-organization of filament actin as well as increase in cell proliferation and migration19. The expression of the Snail2 signalling molecule was elevated, suggesting that ZIP8 regulated the expression of Snail219.
Human Cell Lines
SLC39A8 knockout HAP1 cells
SLC39A8 knockout HAP1 cells show a distinct morphological change, reduced F-actin polymerization, and upregulation of E-cadherin19. In this cell line, it has been shown that ZIP8 regulates NF-kB p65 nucleic translocation in cell culture. It was also determined that ZIP8 regulates Snail2, a transcription factor that plays a role in epithelial–mesenchymal transition19.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including EDEM3-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Publications
SLC39A8-CDG Scientific Articles on PubMed
Additional Resources
SLC39A8-CDG on FCDGC
IEMbase
OMIM
Orphanet
Genetic Testing Registry
ClinVar
NIH
GeneCards
UniProt
References
- Park, J. H. et al. SLC39A8 Deficiency: A Disorder of Manganese Transport and Glycosylation. The American Journal of Human Genetics 97, (2015).
- Boycott, K. M. et al. Autosomal-Recessive Intellectual Disability with Cerebellar Atrophy Syndrome Caused by Mutation of the Manganese and Zinc Transporter Gene SLC39A8. The American Journal of Human Genetics 97, (2015).
- Riley, L. G. et al. A SLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. Journal of Inherited Metabolic Disease 40, (2017).
- Park, J. H. et al. N‐glycome analysis detects dysglycosylation missed by conventional methods in <scp>SLC39A8</scp> deficiency. Journal of Inherited Metabolic Disease 43, (2020).
- Bonaventura, E. et al. Clinical, molecular and glycophenotype insights in SLC39A8-CDG. Orphanet Journal of Rare Diseases 16, (2021).
- Choi, E.-K., Nguyen, T.-T., Gupta, N., Iwase, S. & Seo, Y. A. Functional analysis of SLC39A8 mutations and their implications for manganese deficiency and mitochondrial disorders. Scientific Reports 8, (2018).
- Nebert, D. W. & Liu, Z. SLC39A8 gene encoding a metal ion transporter: discovery and bench to bedside. Human Genomics 13, (2019).
- Riley, L. G. et al. A SLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. Journal of Inherited Metabolic Disease 40, (2017).
- Park, J. H. et al. SLC39A8 deficiency: biochemical correction and major clinical improvement by manganese therapy. Genetics in Medicine 20, 259–268 (2018).
- Clayton, P. T. Inherited disorders of transition metal metabolism: an update. Journal of Inherited Metabolic Disease 40, 519–529 (2017).
- Bieberich, E. Synthesis, Processing, and Function of N-glycans in N-glycoproteins. in (2014). doi:10.1007/978-1-4939-1154-7_3.
- Foulquier, F. & Legrand, D. Biometals and glycosylation in humans: Congenital disorders of glycosylation shed lights into the crucial role of Golgi manganese homeostasis. Biochimica et Biophysica Acta (BBA)-General Subjects 129674 (2020) doi:10.1016/j.bbagen.2020.129674ï.
- Lau, K. S. & Dennis, J. W. N-Glycans in cancer progression. Glycobiology vol. 18 (2008).
- Mealer, R. G. et al. The schizophrenia risk locus in SLC39A8 alters brain metal transport and plasma glycosylation. Scientific Reports 10, 13162 (2020).
- SLC39A8-congenital disorder of glycosylation | Rare Diseases Clinical Research Network. https://www.rarediseasesnetwork.org/fcdgc/slc39a8.
- Park, J. H. & Marquardt, T. Treatment Options in Congenital Disorders of Glycosylation. Frontiers in Genetics 12, (2021).
- Dickinson, M. E. et al. High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514 (2016).
- Lin, W. et al. Hepatic metal ion transporter ZIP8 regulates manganese homeostasis and manganese-dependent enzyme activity. Journal of Clinical Investigation 127, 2407–2417 (2017).
- Geng, X. et al. Role of ZIP8 in regulating cell morphology and NF-κB/Snail2 signaling. Metallomics 10, (2018).