
Lay Summary
MAN1B1-CDG is a rare inherited condition that affects many parts of the body. It is a relatively common congenital disorder of glycosylation (CDG) with more than 40 patients reported in the medical literature to date. MAN1B1-CDG is classified as a disorder of N-linked protein glycosylation. MAN1B1-CDG is caused when an individual has mutations in both copies of their MAN1B1 gene, which provides instructions for making an enzyme that helps ensure that misfolded N-linked glycoproteins are broken down. Mutations in the MAN1B1 gene cause proteins to have altered patterns in the sugar chains on N-linked glycoproteins and allows some misfolded proteins to accumulate instead of being degradation. Symptoms of MAN1B1-CDG usually begin at infancy and affect multiple systems. The most common symptoms are developmental delay, low muscle tone, mild intellectual disability, obesity, mild facial abnormalities, and behavioral problems. Several screening tests are available for MAN1B1-CDG, but a definitive diagnosis is achieved through genetic testing. There are currently no approved treatments for MAN1B1-CDG, and treatment is focused on the management of specific symptoms and preventing disease complications.
Overview
Endoplasmic reticulum mannosyl-oligosaccharide alpha-1,2-mannosidase congenital disorder of glycosylation (MAN1B1-CDG) is a rare autosomal recessive genetic disorder1,2. The first cases of MAN1B1-CDG were reported in 2011, and since then approximately 40 cases have been described to-date3.
The MAN1B1 gene encodes a protein (MAN1B1) that is thought to play several roles in N-glycoprotein quality control. It is a mannosidase and can catalyze the removal of a specific mannose sugar from the glycan chain of recently synthesized N-glycoproteins (Figure 1) that are improperly folded1. Removal of this mannose residue marks them for degradation through ER-associated degradation (ERAD). As well, if misfolded N-glycoproteins escape ERAD and enter the Golgi, MAN1B1 can bind to them, preventing their secretion and helping to shuttle them back to the ER for degradation4,5. Deficiency in MAN1B1 can cause misfolded proteins to accumulate, which can cause stress and alterations to the Golgi structure1. Due to the important role the Golgi plays in N-glycan processing, this leads to altered glycosylation patterns for some N-glycoproteins and causes N-linked CDG (Type 2).
Symptoms begin in infancy and typically include developmental delay, low muscle tone, mild intellectual disability, obesity, mild facial abnormalities, and behavioral problems3,6–8. Definitive diagnosis can only be achieved through genetic testing, but transferrin analysis shows a characteristic Type 2 pattern3,8,9. Currently, there is no cure or treatment for MAN1B1-CDG, and therapy is based on management of symptoms.
Synonyms
- Carbohydrate deficient glycoprotein syndrome Type 2 due to MAN1B1 deficiency
- Congenital disorder of glycosylation type 2 due to MAN1B1 deficiency; Congenital disorder of glycosylation Type 2 due to MAN1B1 deficiency
- MAN1B1 deficiency
- Congenital disorder of glycosylation IIu
- Intellectual disability-truncal obesity syndrome
- Rafiq syndrome, RAFQS
Inheritance
MAN1B1-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The MAN1B1 gene encodes the enzyme endoplasmic reticulum mannosyl-oligosaccharide alpha-1,2-mannosidase (MAN1B1), which is also called ERManI. Although the function of the protein remains incompletely understood, it is believed that MAN1B1 is involved in the quality control process for the synthesis of N-linked glycoproteins in two ways:
- Initiating the degradation of misfolded N-glycoproteins through ERAD by removing a specific mannose sugar from the N-glycan
- Binding to misfolded N-glycoproteins that escape the ER and enter the Golgi, preventing their secretion and shuttling them back to the ER for degradation
MAN1B1 possesses mannosidase activity and can catalyze the removal of the middle mannose mannose sugar from the glycan chain of recently synthesized but improperly folded N-glycoproteins. Removal of this mannose residue marks proteins for degradation through ERAD. There is evidence that other proteins may catalyze the same reaction as MAN1B1, but MAN1B1 is also able to bind to improperly folded N-glycoproteins and prevent their secretion from the Golgi4,5,10.
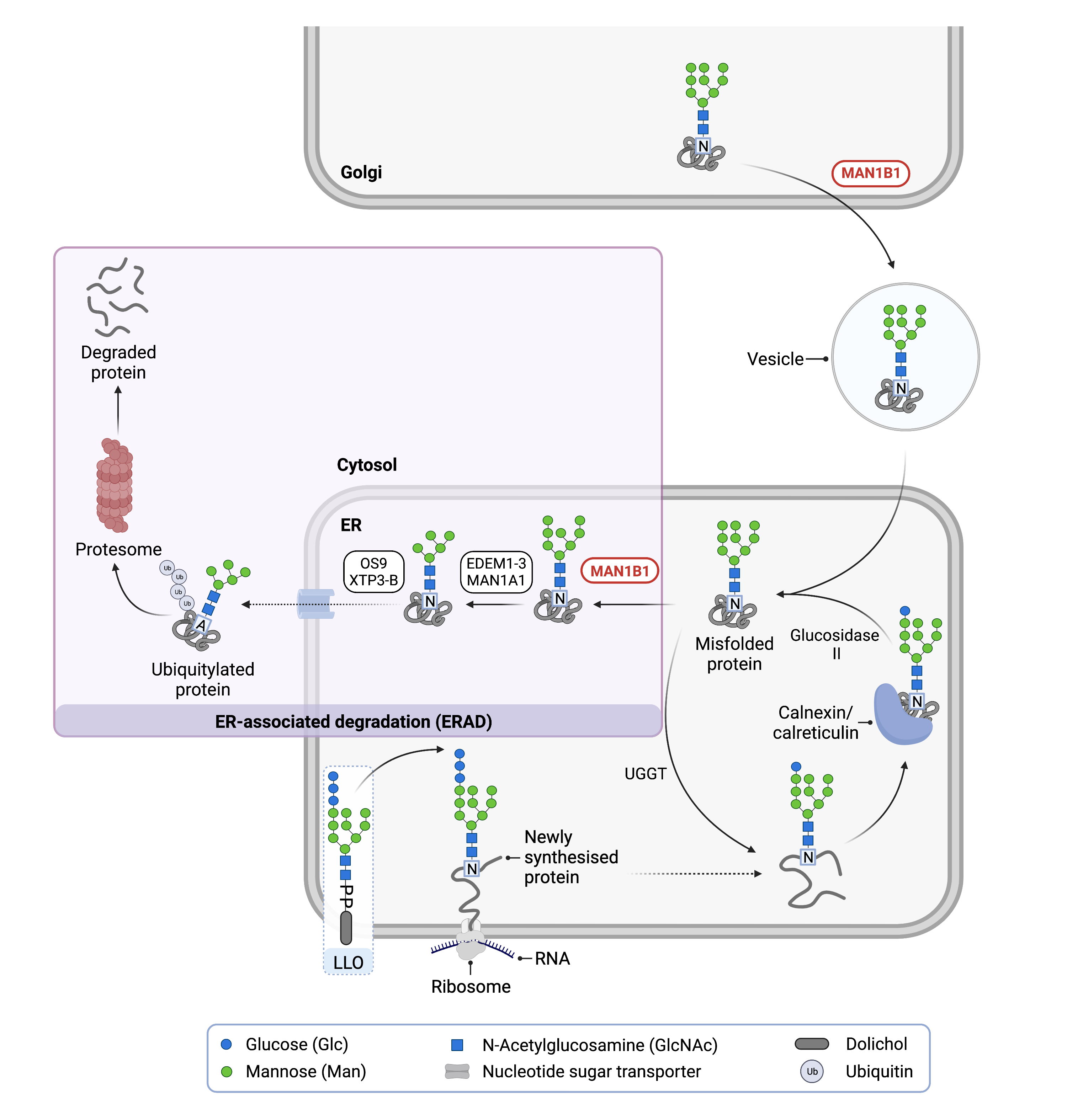
Figure 1: Role of MAN1B1 in Glycosylation.
MAN1B1 is involved in N-glycoprotein quality control. In the ER, MAN1B1 removes the middle mannose from an N-glycan of a misfolded protein, which is needed for protein degradation via the ER-associated degradation (ERAD) pathway. In the Golgi, MAN1B1 shuttles misfolded N-glycoproteins back into the ER for degradation via ERAD.
Role in ERAD
N-linked glycosylation is the process by which a glycan chain is added to the amino acid asparagine on some recently synthesized proteins. N-glycoprotein synthesis is split into three main phases:
- construction of a 14-sugar glycan chain on the lipid dolichol (also known as the lipid-linked oligosaccharide) in the cytoplasm and ER
- transfer of this glycan onto an asparagine residue of a newly synthesized protein in the ER to form an N-glycoprotein
- further trimming of the now N-linked glycan chain occurs as the protein moves from the ER through the Golgi to create a mature N-glycoprotein.
The third step of glycosylation also incorporates a quality control process. After attachment of the glycan to the polypeptide (step 2), the three glucose residues are sequentially removed from the glycan chain by glycosidases, and the resultant N-glycoprotein undergoes additional folding with the help of chaperones. If the protein is folded correctly, removal of the terminal glucose is a signal for the N-glycoprotein to be transported to the Golgi apparatus. However, if the protein is not folded correctly, the enzyme UDP-glucose:glycoprotein glucosyltransferase (UGGT) will re-add a glucose molecule to the glycan chain11. Next, two chaperones, calreticulin and calnexin, bind to the misfolded-protein and prevent it from leaving the ER and giving it more time to become properly folded12–14. If an N-glycoprotein is unable to fold correctly, it will eventually be marked for degradation through ERAD.
The first step of ERAD is the removal of a mannose residues from the glycan chain of a misfolded N-glycoprotein by MAN1B1. This allows other mannosidases to trim addition mannoses from the glycan. The now-truncated N-glycoprotein is then translocated into the cytosol where it has multiple ubiquitin molecules added to the protein, a process called polyubiquitination. The ubiquitination marks the misfolded glycoprotein for transport to the proteasome, where it is chemically broken apart.
Non-catalytic Role in Proteostasis
Besides its ability to initiate the degradation of misfolded glycoproteins by ERAD, MAN1B1 in the Golgi can bind to misfolded proteins that have escaped the ER. MAN1B1 then helps shuttle them back to the ER, where they have the opportunity to undergo ERAD (Figure 2)1,4,5. This binding of MAN1B1 to misfolded proteins in the Golgi occurs whether or not the protein has a glycan chain. Thus, MAN1B1 exerts a positive effect on proteostasis catalytically through its mannosidase activity in the ER, and non-catalytically by binding to misfolded proteins in the Golgi.
Disease Mechanism
Mutations in the MAN1B1 gene lead to the production of an abnormal enzyme with reduced or no activity, and results in the accumulation of some misfolded N-glycoproteins and N-glycoproteins with abnormal glycan structures3,8,14,15. Structural changes to the Golgi, possibly due to the inadequate degradation of misfolded proteins, is also noted in MAN1B1-CDG; this may impact the normal processing of N-glycan chains on otherwise normally folded proteins1. Inadequate MAN1B1 activity on misfolded N-glycoproteins may result in inappropriate trimming of mannose sugars from the N-glycan by other mannosidase enzymes present in the ER and Golgi, resulting in the formation of proteins with abnormal N-glycan chains3,8,14,15.
Mutations
The MAN1B1 gene is found on Chromosome 9 (9q34.3) and 19 mutations have been reported for the MAN1B1 gene including 11 missense mutations, 1 nonsense mutation, 5 deletion mutations, and 2 splice variants, but the clinical importance of most of these mutations remains unclear6.
Signs & Symptoms
Clinical Presentation
Individuals with MAN1B1-CDG typically develop signs and symptoms during infancy. MAN1B1-CDG is primarily characterized by variable intellectual disability and intellectual delay, mild abnormal facial features (facial dysmorphisms), low muscle tone (hypotonia), and obesity particular around the abdomen (truncal obesity)1–3,6,7,9,16. The characteristic clinical presentations of MAN1B1-CDG include:
- Neurologic – developmental delay, intellectual deficiency, delayed speech development, and low muscle tone that may cause excessive drooling. Brain MRI may appear abnormal in some patients. Intermittent seizures are an uncommon symptom
- Feeding/Growth – patients are often significantly overweight with truncal obesity
- Muscoloskeletal – highly flexible joints (joint hyperlaxity), loose skin, sunken-in chest, scoliosis, and long, thin fingers
- Abnormal Features – facial features may include down-slanting opening between the eyelids (palpebral fissures), prominent eyebrows with lateral thinning, bulbous tip of the nose, large ears, and tent-shaped mouth with thin upper lip
- Behavioral Problems – verbal and physical aggression, overeating, and/or inappropriate sexual behaviour
Biochemical Abnormalities
MAN1B1-CDG does not have many significant biochemical markers, but elevated liver enzymes (transaminases) and/or abnormal blood clotting (coagulation) tests may be found in some patients3,9.
Diagnosis
MAN1B1-CDG should be suspected in any multisystem disorder, especially when developmental delay, hypotonia, and facial dysmorphism is present. Screening in suspected patients typically begins with a blood test to analyze serum transferrin. Profiling of total serum N-glycans by mass spectrometry can also be performed3,8,15,17. However, direct molecular genetic testing is the only way to definitively diagnose MAN1B1-CDG.
Transferrin Analysis
Affected individuals will display a Type 2 pattern on transferrin isoelectric focusing (TIEF), with markedly increased trisialotransferrin and decreased tetrasialotransferrin. Type 2 patterns are observed in N-linked CDG that involve processes that occur after the oligosaccharide has been attached to the protein.
Total Serum N-Glycan Analysis
In MAN1B1-CDG, accumulation of abnormal N-glycan structures only occurs in trace amounts and analysis of the overall glycan pool in human serum is challenging. However, treatment of whole human serum glycoproteins with Endo-beta-N-acetylglucosaminidase H (Endo H) causes the release of only high-mannose and hybrid glycan structures15. The analysis of these glycans by MALDI mass spectrometry reveals hybrid glycan structures uniquely enriched in MAN1B1-CDG.
Biomarkers
There are no currently no known biomarkers specific to MAN1B1-CDG.
Classification
MAN1B1-CDG is classified as a disorder of N-linked protein glycosylation. Under the former CDG classification system, MAN1B1-CDG is classified as a Type 2 CDG, which arise due to defects in the processing of N-glycans attached to proteins.
Prognosis
Information on long-term outcomes for MAN1B1-CDG patients is unavailable, but patients in their 20s have been reported. Developmental delays reported include delayed walking until 4 years old, delayed speech development and limited speech after development and assistance required for bathing and dressing2.
Management
Management of MAN1B1-CDG requires a multidisciplinary team and may include combinations of physical therapy, occupational therapy, oral motor and speech therapy, and psychiatry. Excessive drooling due to oral-facial hypotonia may be managed with subcutaneous scopolamine injections3.
Therapies
There are currently no treatment options available for MAN1B1-CDG. Disulfiram can reduce protein translation and potentially reduce the amount of misfolded glycoproteins formed; oral disulfiram was tested in one MAN1B1-CDG patients but no improvement to transferrin glycosylation was observed17. Treatment is focused on management of symptoms and prevention of complications.
Work being done at JDC Research Co is looking at developing gene-based therapies and small molecule therapies to find a treatment or cure for MAN1B1-CDG18.
Research Models
Model organisms for MAN1B1 are currently in development to study MAN1B1-CDG. Several human cell lines have been used to investigate the role MAN1B1 plays in protein quality control.
Fly (D. melanogaster)
Dr. Clement Chow and JDC Research Co have generated a Man1b1 Drosophila model to study gene function through a knockdown mutation in the eye, which is not lethal and makes the model suitable for drug screen studies. In addition, JDC Research Co in partnership with Vivian Therapeutics generated temperature sensitive knockdowns of Man1b1, which have shown that flies with reduced levels of Man1B1 protein have a decreased survival rate to adulthood18.
Human Cell Lines
Patient derived fibroblasts
Man1b1 patient derived fibroblasts have shown abnormal Golgi morphology, where Golgi compartments are disorganized and fragmented1. However, work done by JDC Research Co found a more condensed signal of Golgi markers (TGN6 and WGA) around the nucleus in patient cells18.
Human embryonic kidney (HEK293T) MAN1B1 knockout cells
MAN1B1 knockout HEK293T cells were prepared and then transfected to express different mutated forms of MAN1B1 to study their effect on the fate of three simultaneously expressed misfolded N-glycoproteins5. The system identified the cytoplasmic chain of MAN1B1 as being important for the non-enzymatic recognition of misfolded N-glycoproteins.
HeLa immortal cell line
HeLa cells were used to further demonstrate the non-enzymatic role MAN1B1 can play as a gatekeeper in proteostasis4.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including MAN1B1-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Groups
Publications
MAN1B1-CDG Scientific Articles on PubMed
Additional Resources
MAN1B1-CDG on FCDGC
MAN1B1-CDG Infographic
MAN1B1-CDG Clinical Utility Gene Card
IEMbase
GeneReviews
OMIM
OrphaNet
GARD
Genetic Testing Registry
ClinVar
NIH
GeneCards
UniProt
References
- Rymen, D. et al. MAN1B1 deficiency: an unexpected CDG-II. PLoS Genet 9, (2013).
- Rafiq, M. A. et al. Mutations in the Alpha 1,2-Mannosidase Gene, MAN1B1, Cause Autosomal-Recessive Intellectual Disability. American Journal of Human Genetics 89, 176 (2011).
- Sakhi, S. et al. MAN1B1-CDG: Three new individuals and associated biochemical profiles. Mol Genet Metab Rep 28, (2021).
- Iannotti, M. J., Figard, L., Sokac, A. M. & Sifers, R. N. A Golgi-localized mannosidase (MAN1B1) plays a non-enzymatic gatekeeper role in protein biosynthetic quality control. J Biol Chem 289, 11844–11858 (2014).
- Sun, A. H., Collette, J. R. & Sifers, R. N. The cytoplasmic tail of human mannosidase Man1b1 contributes to catalysis-independent quality control of misfolded alpha1-antitrypsin. Proc Natl Acad Sci U S A 117, 24825–24836 (2020).
- Jaeken, J., Lefeber, D. J. & Matthijs, G. Clinical utility gene card for: MAN1B1 defective congenital disorder of glycosylation. European Journal of Human Genetics 2016 24:7 24, 1–3 (2015).
- Balasubramanian, M. & Johnson, D. S. MAN1B-CDG: Novel variants with a distinct phenotype and review of literature. European Journal of Medical Genetics 62, 109–114 (2019).
- van Scherpenzeel, M. et al. Diagnostic serum glycosylation profile in patients with intellectual disability as a result of MAN1B1 deficiency. Brain 137, 1030–1038 (2014).
- Kasapkara, C. S. et al. MAN1B1-CDG: novel patients and novel variant. J Pediatr Endocrinol Metab 34, 1207–1209 (2021).
- George, G. et al. EDEM2 stably disulfide-bonded to TXNDC11 catalyzes the first mannose trimming step in mammalian glycoprotein ERAD. Elife 9, (2020).
- Dejgaard, S., Nicolay, J., Taheri, M., Thomas, D. Y. & Bergeron, J. J. M. The ER Glycoprotein Quality Control System. Current Issues in Molecular Biology 2004, Vol. 6, Pages 29-42 6, 29–42 (2003).
- Gañán, S., Cazzulo, J. J. & Parodi, A. J. A major proportion of N-glycoproteins are transiently glucosylated in the endoplasmic reticulum. Biochemistry 30, 3098–3104 (1991).
- Michalak, M., Mariani, P. & Opas, M. Calreticulin, a multifunctional Ca2+ binding chaperone of the endoplasmic reticulum. Biochem Cell Biol 76, 779–785 (1998).
- Hammond, C., Braakman, I. & Helenius, A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc Natl Acad Sci U S A 91, 913 (1994).
- Duvet, S. et al. Use of Endoglycosidase H as a diagnostic tool for MAN1B1-CDG patients. Electrophoresis 39, 3133–3141 (2018).
- Hoffjan, S., Epplen, J. T., Reis, A. & Abou Jamra, R. MAN1B1 Mutation Leads to a Recognizable Phenotype: A Case Report and Future Prospects. Mol Syndromol 6, 58–62 (2015).
- Kemme, L. et al. Translational balancing questioned: Unaltered glycosylation during disulfiram treatment in mannosyl-oligosaccharide alpha-1,2-mannnosidase-congenital disorders of glycosylation (MAN1B1-CDG). JIMD Rep 60, 42–55 (2021).
- Our Research — JDC Research Co. https://www.jdcresearchco.com/ourresearch.