
Lay Summary
SRD5A3-CDG, formerly known as CDG-Iq, is a rare inherited condition that affects several body systems. To date, approximately 50 cases of SRD5A3-CDG have been reported in the literature. SRD5A3-CDG is classified as a disorder of multiple glycosylation pathways and can be subcategorized as a disorder of dolichol metabolism. It is caused when an individual has mutations in both copies of the SRD5A3 gene, which provides instructions for making an enzyme that converts the molecule, polyprenol, into dolichol – a lipid that is needed for different types of glycosylation. Mutations in the SRD5A3 gene reduce the availability of dolichol, leading to proteins and lipids with abnormal sugar chains. Symptoms of SRD5A3-CDG begin in infancy and commonly include neurological abnormalities (problems with balance and coordination, low muscle tone and intellectual disability) vision loss early on in life, eye abnormalities, unusual facial features and skin symptoms. Some individuals may present with heart and liver problems and feeding difficulties. A screening test is available for SRD5A3-CDG but a definitive diagnosis is achieved through genetic testing. There are currently no approved treatments for SRD5A3-CDG but gene therapy and drug repurposing are currently being explored in pre-clinical studies. Current treatment focuses on managing specific symptoms and preventing complications.
Overview
Steroid 5ɑ-reductase type 3 congenital disorder of glycosylation (SRD5A3-CDG) is a rare autosomal recessive genetic disorder. The first genetically confirmed case of SRD5A3-CDG was reported in 2010 1,2. To date approximately 50 cases with molecular confirmed diagnosis have been reported in the literature—although there are several cases that have not be yet been reported1–14.
The SRD5A3 gene encodes polyprenol reductase, an enzyme that catalyzes the conversion of the molecule, polyprenol, to the lipid, dolichol1. Biosynthesis of dolichol is one of the first steps in glycosylation, where dolichol acts as a lipid carrier for the glycan precursor in the early stages of N-glycosylation of proteins. In addition, dolichol is also a carrier for monosaccharides used as donors in O-mannosylation, C-mannosylation and GPI anchor biosynthesis. Mutations in the SRD5A3 gene results in hypoglycosylation of N-glycoproteins and may cause defects in multiple glycosylation pathways.
Symptoms of SRD5A3-CDG begin in infancy and typically include neurological abnormalities, such as ataxia, hypotonia, a small cerebellum and intellectual disability, earl-onset visual impairment and variable ophthalmological abnormalities, facial dysmorphisms, and skin abnormalities. Symptoms that may develop over time include curving of the spine, cataracts, and retinitis pigmentosa2,4,11,12. Definitive diagnosis can only be achieved through genetic testing, but transferrin analysis typically shows a Type I pattern5. Although there are currently no approved treatments for SRD5A3-CDG, gene therapy and drug repurposing are currently under pre-clinical investigation.
Synonyms
- CDG-Iq; CDG-1q
- SRD5A3-deficiency
- Steroid 5α-reductase 3 deficiency
- SRD5A3-congenital disorder of glycosylation
- Polyprenol reductase deficiency
- Ocular coloboma with ichthyosis, brain malformations and endocrine abnormalities
Inheritance
SRD5A3-CDG is inherited in an autosomal recessive fashion, with one mutation inherited from each asymptomatic (carrier) parent.
Gene Function
The SRD5A3 gene encodes an enzyme called polyprenol reductase (SRD5A3) it has roles in both glycosylation1,15 and androgen biosynthesis16. In glycosylation, polyprenol reductase is needed to generate dolichol, which is used as a lipid carrier of the oligosaccharide precursor in N-glycosylation and as a donor of glucose and mannose for several glycosylation pathways.
Dolichol Biosynthesis
Dolichol is a lipid that can be found in the membrane of the ER and is important for glycosylation in two ways: it acts a lipid carrier for the 14-sugar oligosaccharide that is transferred onto nascent proteins during N-glycosylation and is a monosaccharide donor of mannose and glucose sugars for N-glycosylation, O- and C-mannosylation and GPI anchor biosynthesis (Figure 1)17.
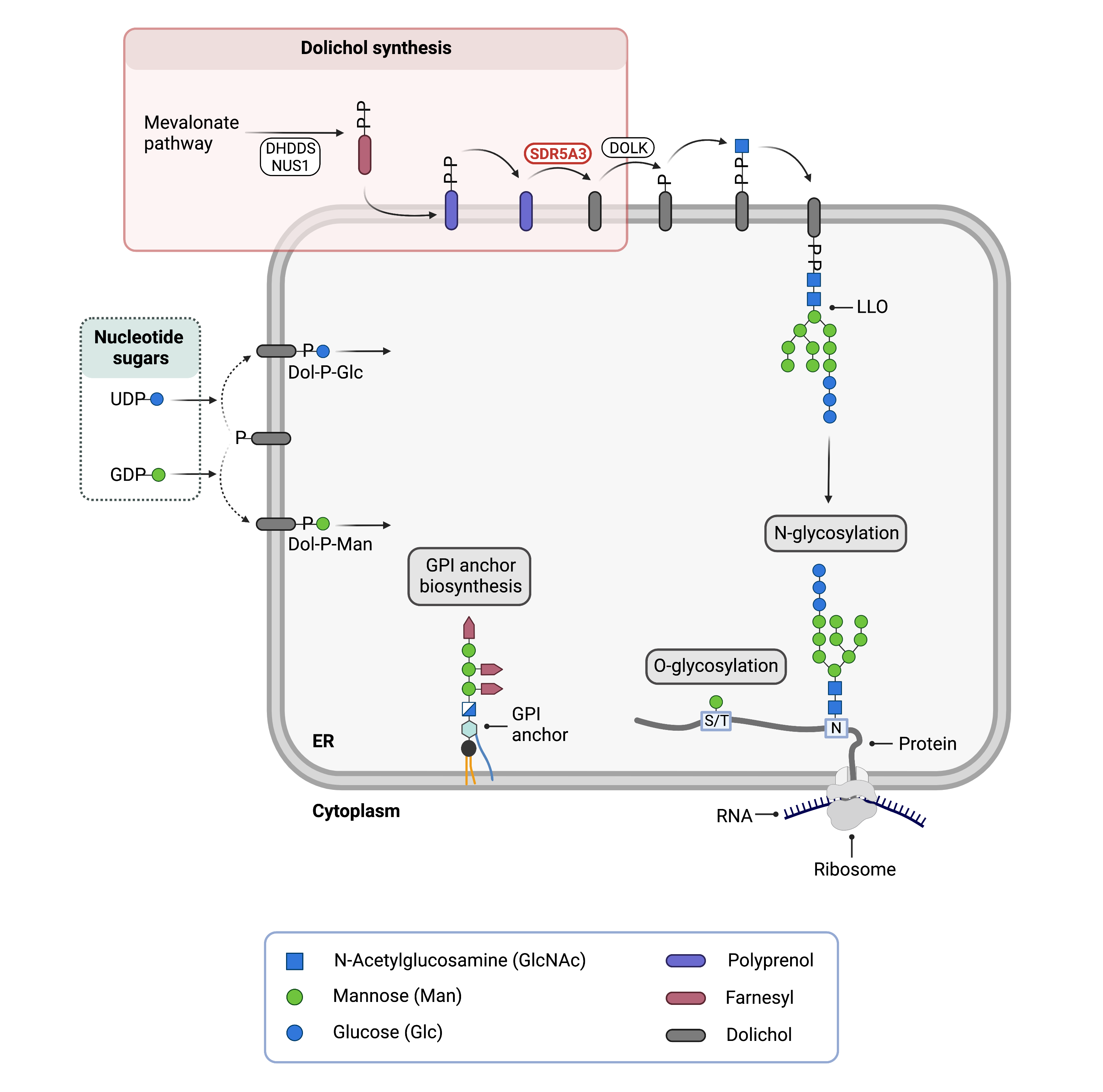
Figure 1. Role of SRD5A3 in glycosylation.
SRD5A3 is an enzyme that is needed for dolichol synthesis, where it catalyzes the conversion of polyprenol into dolichol. In N-glycosylation, the oligosaccharide precursor is built upon phosphorylated dolichol (together forming the lipid-linked oligosaccharide; LLO) and the 14-sugar oligosaccharide is transferred onto asparagine (N) residues of newly synthesized proteins in the ER lumen. Phosphorylated dolichol is also a donor of glucose (Dol-P-Glc) and mannose (Dol-P-Man) sugars used in multiple glycosylation pathways: N-glycosylation, O-glycosylation and GPI anchor biosynthesis.
Dolichol is synthesised through a series of enzymatic reactions in the cytosol and ER membrane. SRD5A3 catalyzes the final step in dolichol biosynthesis which takes place in the ER membrane– the conversion of polyprenol to dolichol1. Once dolichol is generated, it must be phosphorylated by the enzyme dolichol kinase (DOLK), generating dolichol-phosphate (Dol-P) before it can be used as a lipid carrier or monosaccharide donor.
In N-glycosylation, Dol-P acts as a lipid carrier upon which the 14-sugar oligosaccharide, GlcNAc2Man9Glc3, is built. Together, phosphorylated dolichol (Dol-PP) and the oligosaccharide form the lipid-linked oligosaccharide (LLO). Once assembly of the LLO is complete, the oligosaccharide component is transferred onto an asparagine residue of a newly synthesized protein the ER.
Sugar donors Dol-P-mannose and Dol-P-glucose are generated upon transfer of mannose and glucose sugars from nucleotide sugars GDP-mannose and UDP-glucose, respectively. These dolichol-linked sugars act as donors of mannose and glucose for multiple glycosylation pathways.
Androgen Biosynthesis
SRD5A3 belongs to the 5α-reductase family of enzymes which play a critical role in the biosynthesis of androgens – hormones which contribute to growth and development. In addition to its role in dolichol synthesis, SRD5A3 catalyzes the conversion of the androgen testosterone (T) into another androgen, 5-alpha-dihydrotestosterone (DHT)16.
Disease Mechanism
Mutations in the SRD5A3 gene can the production of an abnormal enzyme that has little or no activity, reducing the amount of dolichol available for glycosylation. In turn, the amount of LLO synthesized is reduced, resulting in hypoglycosylation of N-glycoproteins1,18. As phosphorylated dolichol acts as a donor of glucose and mannose residues, insufficient levels of dolichol may disrupt multiple glycosylation pathways and lead to abnormal glycans on proteins and lipids. Additionally, elevated levels of polyprenol have been observed in patient samples1,8, however it has not been determined whether this plays a role in the pathology of SRD5A3-CDG.
Early-onset retinal dystrophy is a defining clinical feature of SRD5A3-CDG and it has been hypothesized that abnormal N-glycosylation of rhodopsin (a pigment in the retinal that is essential for vision) may in part be causative4,12,14. As early visual impairment is clinical feature of several other CDG caused by defects in the dolichol metabolism19–21, it is thought that dolichol plays an important role in the normal development of the eyes. Lastly, hypoglycosylation of specific proteins in the brain may disrupt normal development of the cerebellum, resulting motor impairment in patients15.
Mutations
The SRD5A3 gene is located on Chromosome 4 (4q13) and 24 pathogenic mutations have been reported in the literature including seven different nonsense variants, six missense variants, eight frameshift insertions/deletions, one splice site mutation, one complex genomic rearrangement and one copy number variant3–6,14. The homozygous mutation c.57G > A (p.Trp19Ter) has been reported in nearly half of all cases documented to date is associated with early-onset retinal dystrophy in patients4,12.
Biallelic pathogenic variants in SRD5A3 are also known to cause Kahrizi syndrome, a phenotypically similar disorder that is characterized by coloboma, cataract, kyphosis, and intellectual disability22,23.
Signs & Symptoms
Clinical Presentation
Individuals with SRD5A3-CDG typically develop signs and symptoms during infancy. SRD5A3-CDG is primarily characterized by early-onset severe visual impairment and various eye abnormalities, cerebellar ataxia, low muscle tone, developmental disability and skin anomalies1–14. The characteristic clinical presentation of SRD5A3-CDG includes:
- Neurological symptoms – poor muscle tone (hypotonia), lack of muscle control and coordination (ataxia), psychomotor disability, developmental delay, midline brain malformation, underdeveloped cerebellum (cerebellar atrophy)
- Ophthalmological symptoms– severe visual impairment at a young age (early-onset retinal dystrophy), involuntary eye movement (nystagmus), coloboma, underdeveloped optic disk and nerve
- Skin symptoms – dark patches (hyperpigmentation), dry skin, excessive hair growth, accumulation of dead skin cells (ichthyosis), loose skin, thickening of the skin on palms of hands and soles of feet
- Facial dysmorphisms – arched eyebrows, wide eyes, shallow nasal bridge, short nose, and large mouth
Symptoms that may develop over time are curving of the spine (kyphosis), cataracts and retinal degeneration (retinitis pigmentosa)5. Fatty liver disease (hepatic steatosis) has also been reported in one patient14.
Symptoms reported in some patients include feeding problems, gastrointestinal problems, heart abnormalities, dystonia, swelling of the liver and spleen, joint hypermobility and anxiety disorder4,5.
Biochemical Abnormalities
Biochemical abnormalities include elevated liver enzymes (transaminases), underactive thyroid (hypothyroidism), and reduced levels of blood clotting factors antithrombin and protein C5.
Classification
SRD5A3-CDG is classified as a disorder of multiple glycosylation pathways and within this group, a disorder of dolichol biosynthesis.
Under the former classification system, SRD5A3-CDG is classified as a Type I CDG, which arise due to defects in the synthesis or transfer of lipid-linked oligosaccharides to N-glycoproteins.
Diagnosis
SRD5A3-CDG should be suspected in patients presenting with early-onset visual impairment and variable eye abnormalities (coloboma, cataracts), neurological symptoms (cerebellar ataxia, intellectual disability, developmental delay) and skin symptoms 4. Screening in suspected patients begins with analysis of serum transferrin isoforms with patients often displaying a type I pattern, however, a definitive diagnosis can only be achieved through genetic testing.
Transferrin Analysis
Affected individuals will typically show a type I pattern by transferrin isoelectric focusing (TIEF)5. However, normal transferrin patterns have been observed in several patients4,7,11,24.
Biomarkers
High Mannose N-Glycans
A high abundance of high mannose Man3 (Man3GlcNAc2) N-glycans and high ratio of Man3:Man4 glycans has been observed in plasma samples from a cohort SRD5A3-CDG patients which may serve as a diagnostic biomarker18.
Prognosis
Prognosis depends on severity of an individual’s symptoms, with some reported cases not being diagnosed until adulthood (30s-40s)10. Individuals affected with SRD5A3-CDG typically develop eye abnormalities that progress as they age, such as cataracts, glaucoma, retinal degeneration, leading SRD5A3-CDG patient eye health to be monitored4,11. Affected individuals may also develop a curved spine with age11.
Management
Management of SRD5A3-CDG focuses on managing symptoms and often includes a combination of physical therapy, occupational therapy, and speech or vision therapy, as well as palliative measures, requiring a multi-disciplinary team5.
Therapies
There are currently no treatments available for SRD5A3-CDG although several therapies are currently under investigation by Cure SRD5A3 including gene therapy and repurposed approved drugs.
Research Models
There are several available research models of SRD5A3-CDG, including yeast, worm, zebrafish and mouse models, as well as patient-derived fibroblast cell lines.
Yeast (S. cerevisiae)
The SRD5A3 gene is the human ortholog of yeast DFG10. Mutant DFG10 yeast display growth delays and hypoglycosylation of the secreted glycoprotein: Carboxypeptidase Y. Mutants also display defective metabolism of polyprenol to dolichol, evident by elevated polyprenol and reduced dolichol levels1.
Worm (C. elegans)
Nematode B0024.13 is orthologous to human SRD5A3. Two Srd5a3-deficient worm models have been generated by Cure SRD5A3 using CRISPR/Cas9 gene editing: a gene deletion and W6X mutant which corresponds to the most common SRD5A3-CDG patient mutation (W19X). Srd5a3 mutants have a reduced lifespan and display developmental delay, slow motility, reduced progeny numbers and differential expression of ER stress-related genes. Mutant worms also display abnormal levels of several metabolites in the mevalonate pathway, including decreased levels of dolichol.
Fish (D. rerio)
A srd5a3 knockout zebrafish model has been generated by Cure SRD5A3 and is currently being characterized.
Mouse (M. musculus)
Srd5a3-/- constitutive knockout mouse
An Srd5a3 constitutive knockout mouse is embryonic lethal beyond E12.5 (IMPC). Embryos are smaller, displayed open neural tubes and dilated hearts1. Whole transcriptome analysis of Srd5a3 mutants have shown activation of the unfolded protein response, upregulation of genes in the mevalonate pathway and downregulation of genes involved in general metabolic processes and embryonic development programs1. Elevated polyprenol levels have also been detected in this mouse model of Srd5a3 deficiency1.
Cerebellum-specific Srd5a3 conditional knockout mouse
A cerebellum-specific Srd5a3 knockout mouse has been generated11. The phenotype of this model recapitulates some of the neurological symptoms present in SRD5A3-CDG patients, such as impaired motor coordination. Mice also display abnormal cerebellum granule cell development. Proteomic analysis has shown a decrease in abundance of N-glycoproteins that have many N-glycosylation sites, specifically proteins belonging to the immunoglobulin superfamily of cell adhesion molecules (IgSF-CAMs). Several IgSF-CAMs proteins were hypoglycosylated, resulting in impaired IgSF-CAM-mediated neurite outgrowth and axon guidance in the Srd5a3 knockout cerebellum. This model is currently be used to evaluate a gene therapy for SRD5A3-CDG in pre-clinical studies.
Conditional-ready floxed Srd5a3 knockout mouse
A conditional-ready floxed Srd5a3 knockout mouse has been generated (JAX). This mouse model can be bred to different CRE driver mice to investigate Srd5a3 deficiency in tissue-specific and inducible models.
Rod-specific Srd5a3 conditional knockout mouse
A rod-specific conditional knockout mouse is currently being generated by Cure SRD5A3 to study retinal degeneration in SRD5A3-CDG.
Patient-Derived Fibroblasts
SRD5A3-CDG patient-derived fibroblasts display elevated polyprenol levels, normal dolichol levels and variable levels of lipid-linked oligosaccharides1,2,9.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including SRD5A3-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Organizations
Publications
SRD5A3 scientific articles on PubMed
Additional Resources
SRD5A3-CDG on FCDGC
SRD5A3-CDG Clinical Utility Gene Card
IEMbase
OrphaNet
OMIM
GARD
NIH
Gene Cards
Uniprot
Genetic Testing Registry
ClinVar
References
- Cantagrel, V. et al. SRD5A3 Is Required for Converting Polyprenol to Dolichol and Is Mutated in a Congenital Glycosylation Disorder. Cell 142, 203–217 (2010).
- Morava, E. et al. A novel cerebello-ocular syndrome with abnormal glycosylation due to abnormalities in dolichol metabolism. Brain 133, 3210–3220 (2010).
- Ben Ayed, I. et al. SRD5A3-CDG: 3D structure modeling, clinical spectrum, and computer-based dysmorphic facial recognition. Am. J. Med. Genet. Part A 1081–1090 (2021) .
- Jaman, N. K. et al. SRD5A3-CDG: Emerging Phenotypic Features of an Ultrarare CDG Subtype. Front. Genet. 12, 1–11 (2021).
- Jaeken, J., Lefeber, D. J. & Matthijs, G. SRD5A3 defective congenital disorder of glycosylation: clinical utility gene card. Eur. J. Hum. Genet. 28, 1297–1300 (2020).
- Rieger, M. et al. SRD5A3-CDG: Twins with an intragenic tandem duplication. Eur. J. Med. Genet. 104492 (2022).
- Mohamed, M. et al. Normal glycosylation screening does not rule out SRD5A3-CDG. Eur. J. Hum. Genet. 19, 1019 (2011).
- Gründahl, J. E. H. et al. Life with too much polyprenol: Polyprenol reductase deficiency. Mol. Genet. Metab. 105, 642–651 (2012).
- Al-Sarraj, Y. et al. A Novel Missense Mutation in SRD5A3 Causes Congenital Disorder of Glycosylation Type I (Cerebello-Ocular Syndrome). J. Inborn Errors Metab. Screen. 2, 1–5 (2014).
- Kara, B., Ayhan, Ö., Gökçay, G., Başboğaoğlu, N. & Tolun, A. Adult phenotype and further phenotypic variability in SRD5A3-CDG. 1–6 (2014).
- Wheeler, P. G. et al. SRD5A3-CDG: Expanding the phenotype of a congenital disorder of glycosylation with emphasis on adult onset features. Am J Med Genet 170, 3165–3171 (2016).
- Taylor, R. L. et al. Association of steroid 5α-reductase type 3 congenital disorder of glycosylation with early-onset retinal dystrophy. JAMA Ophthalmol. 135, 339–347 (2017).
- Gupta, N. et al. Identification of a case of SRD5A3-congenital disorder of glycosylation (CDG1Q) by exome sequencing. Indian J. Med. Res. 147, 517–520 (2018).
- Kousal, B. et al. Review of SRD5A3 Disease-Causing Sequence Variants and Ocular Findings in Steroid 5α-Reductase Type 3 Congenital Disorder of Glycosylation, and a Detailed New Case. Folia Biologica (Praha) vol. 65 (2019).
- Medina-Cano, D. et al. High N-glycan multiplicity is critical for neuronal adhesion and sensitizes the developing cerebellum to N-glycosylation defect. Elife 7, 1–27 (2018).
- Azzouni, F., Godoy, A., Li, Y. & Mohler, J. The 5 alpha-reductase isozyme family: A review of basic biology and their role in human diseases. Adv. Urol. 2012, (2012).
- Welti, M. Regulation of dolichol-linked glycosylation. Glycoconj. J. 30, 51–56 (2013).
- Abu Baker., N. et al. Synergistic use of glycomics and single-molecule molecular inversion probes (smMIPs) for identification of congenital disorders of glycosylation type-1. J. Inherit. Metab. Dis. (2022).
- Cantagrel, V. & Lefeber, D. J. From glycosylation disorders to dolichol biosynthesis defects: a new class of metabolic diseases. J Inherit Metab Dis (2011).
- Wolfe, L. A., Morava, E., He, M., Vockley, J. & Gibson, K. M. Heritable disorders in the metabolism of the dolichols: A bridge from sterol biosynthesis to molecular glycosylation. Am. J. Med. Genet. Part C Semin. Med. Genet. 160 C, 322–328 (2012).
- Buczkowska, A., Swiezewska, E. & Lefeber, D. J. Genetic defects in dolichol metabolism. J Injerit Metab Dis. 38, 157–169 (2015).
- Kahrizi, K. et al. An autosomal recessive syndrome of severe mental retardation, cataract, coloboma and kyphosis maps to the pericentromeric region of chromosome 4. Eur. J. Hum. Genet. 17, 125–128 (2009).
- Kahrizi, K. et al. Next generation sequencing in a family with autosomal recessive Kahrizi syndrome (OMIM 612713) reveals a homozygous frameshift mutation in SRD5A3. Eur. J. Hum. Genet. 19, 115–117 (2011).
- Al-Gazali, L., Hertecant, J., Algawi, K., El Teraifi, H. & Dattani, M. A new autosomal recessive syndrome of ocular colobomas, ichthyosis, brain malformations and endocrine abnormalities in an inbred Emirati family. Am. J. Med. Genet. Part A 146A, 813–819 (2008).
Show More