
Lay Summary
SLC35A2-congenital disorder of glycosylation (SLC35A2-CDG), formerly known as CDG-IIm, is a rare inherited condition that affects several body systems. To-date, almost 70 cases of SLC35A2-CDG have been reported in the medical literature. SLC35A2-CDG is classified as a disorder of multiple glycosylation pathways and within this category, a disorder of nucleotide sugar synthesis and transport. It is caused by mutations in the SLC35A2 gene found on the X chromosome, which is one of the two sex chromosomes. The SLC35A2 gene provides the instructions for the body to make a protein that transports a high-energy form of the sugar galactose, called a nucleotide sugar, into the Golgi Apparatus (a compartment in the cell that is important for glycosylation). Galactose is important for many glycosylation pathways. Mutations in the SLC35A2 gene limits the amount of galactose available to be added to sugar chains in the Golgi, resulting in shortened chains. Symptoms of SLC35A2-CDG begin at infancy and are primarily characterized by developmental delay, seizures, and low muscle tone. Visual impairments, facial and bone abnormalities, and organ abnormalities may also be present in individuals with this disorder. SLC35A2-CDG is only diagnosed through genetic testing. Currently, galactose supplementation is being tested as a treatment option for SLC35A2-CDG. Present treatment is focused on the management of specific symptoms and preventing complications.
Overview
Solute Carrier Family 35 Member A2 congenital disorder of glycosylation (SLC35A2-CDG) is a rare X-linked dominant genetic disorder. The first reported case of SLC35A2-CDG was in 20131, and there are almost 70 confirmed cases to date, making it one of the more commonly diagnosed CDG2. The SLC35A2 gene encodes the UDP-galactose transporter (UGT), which transports the nucleotide sugar UDP-galactose from the cytoplasm into the Golgi where galactose can be transferred onto oligosaccharide chains on proteins and lipids. As galactose is a component of N- and O-linked glycoproteins, as well as glycosaminoglycans and glycosphingolipids, mutations in SLC35A2 affects multiple types or pathways of glycosylation, resulting in the generation of incomplete oligosaccharides on glycoproteins or glycolipids3.
SLC35A2-CDG is primarily observed in females. Symptoms begin at infancy and the characteristic presentation includes developmental delay, seizures, decreased muscle tone, facial and skeletal abnormalities, visual impairment, and organ abnormalities3. A transferrin analysis can be used to detect SLC35A2-CDG in some cases, however results are often normal in many patients who are no longer in infancy. A definitive diagnosis can only be obtained through genetic testing3–7. No treatment is presently available for treating SLC35A2-CDG, although galactose supplementation is currently being investigated as a therapeutic option for this condition8.
Synonyms
- CDG-IIm
- CDG syndrome type IIm
- Congenital disorder of glycosylation, type IIm
- EIEE22
- Epileptic encephalopathy, early infantile, 22
Inheritance
SLC35A2-CDG is inherited in an X-linked dominant fashion and mainly affects females. These variants arise predominantly from de novo mutations (where the mutation occurs in the egg or sperm cell of a parent or after fertilization) but have been shown to be inherited too9–11. In almost 70 cases diagnosed to date, only 8 males have been diagnosed with SLC35A2-CDG3.
A few males are diagnosed with SLC35A2-CDG displaying mosaicism, where only a proportion of body cells are affected as the mutation is not inherited from the parent. During the early developmental stages in utero, one cell spontaneously acquires a mutation in the SLC35A2 gene, passing the mutationonto its progeny, resulting in some cells carrying the defect12. Having this mutation in all the cells is thought to be lethal13.
Gene Function
The SLC35A2 gene encodes the enzyme UDP-galactose transporter (UGT), which is a nucleotide-sugar transporter. It belongs to the SLC35 family of nucleotide-sugar transporters14–16, which are transmembrane proteins located on the ER and Golgi responsible for translocating nucleotide sugars from the cytosol. The SLC35A2 gene product UGT is an antiporter which transports UDP-galactose from the cytosol into the Golgi in exchange for the reaction by-product called UMP17–19. This galactose residue from UDP-galactose can be added to O-linked oligosaccharide chains or to N-linked glycans undergoing remodelling within the Golgi, meaning mutations can affect glycoprotein, glycolipid, and proteoglycan synthesis20,21.
Transport of UDP-Galactose
For monosaccharides to be added to proteins and lipids, they must first be converted into reactive molecules called nucleotide sugars. Nucleotide sugars are high-energy forms of monosaccharides that are required by enzymes called glycosyltransferases to transfer the monosaccharide to a protein, lipid, or glycan during the process of glycosylation. These high-energy forms of monosaccharides are generated in the cytoplasm and are dependent on a series of enzyme reactions. These sugars are often monosaccharides linked to phosphorylated nucleosides: UDP (uridine diphosphate), GDP (guanosine diphosphate) or CMP (cytidine monophosphate), creating monosaccharide-UDP, -GDP or -CMP, respectively15,22. They are transported into the ER and the Golgi by transporter proteins, called nucleotide sugar transporters.
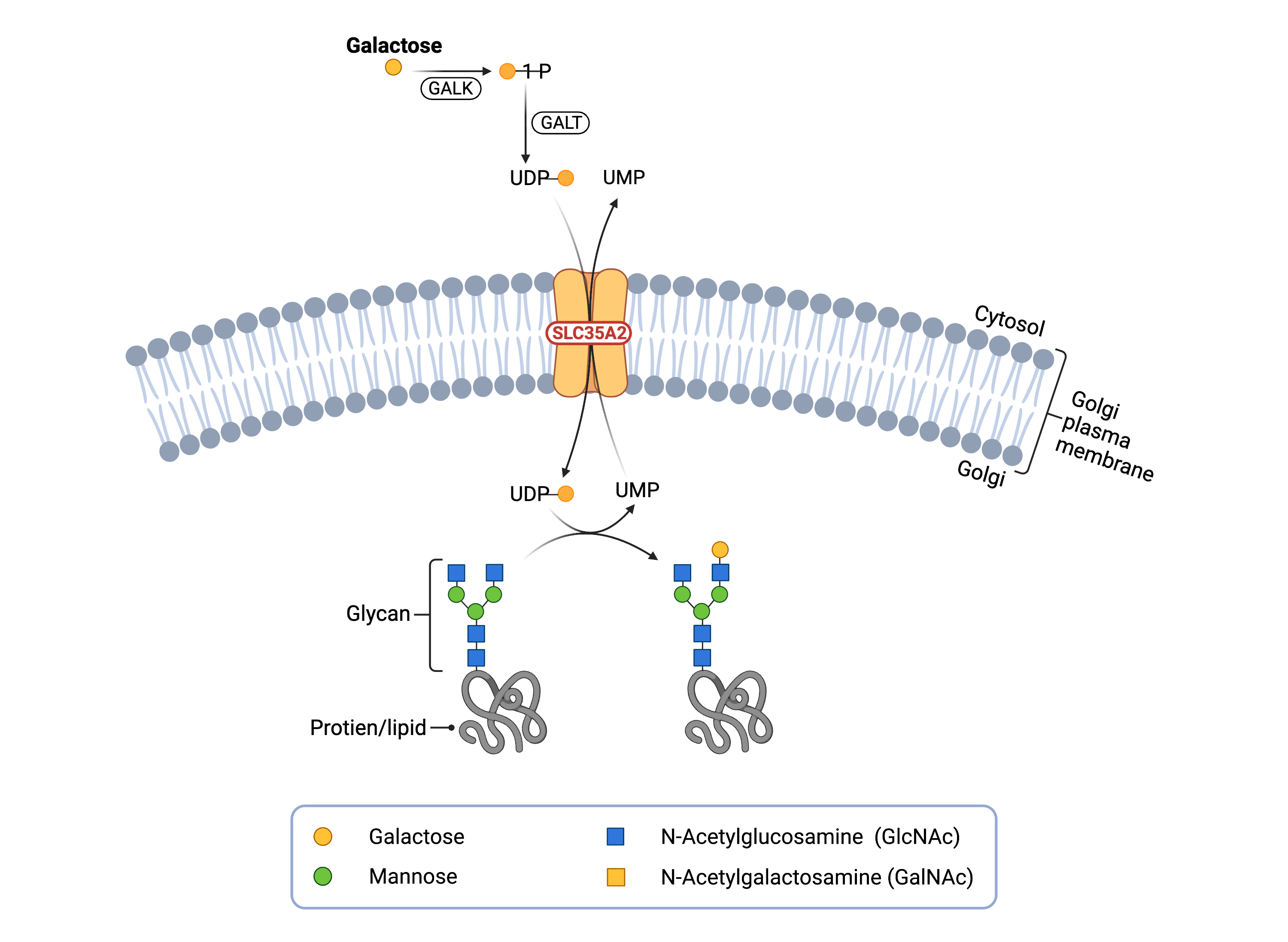
Figure 1. Role of SLC35A2 in glycosylation.
SLC35A2 is a UDP-galactose transporter, which moves UDP-galactose from the cytosol to the Golgi lumen. Galactose can be transferred from UDP-galactose to lipid and protein glycan chains.
Glycosylation occurs inside the ER and the Golgi, which are compartments that are surrounded by membranes that nucleotide sugars cannot pass through. Nucleotide sugar transporters exist in the ER and Golgi membranes to transport nucleotide sugars from the cytosol to the inside of these compartments15. After the initiation of glycosylation in the ER, glycoproteins and glycolipids can be transferred to the Golgi for further modifications, which can include elongation of the sugar chain by the addition of sugars such as galactose21.
The activated form of galactose is the nucleotide sugar UDP-galactose which is generated in the cytosol. UDP-galactose is transported into the Golgi by the enzyme UGT, which is located in the Golgi membrane. Once UDP-galactose has been transported inside the Golgi, galactose-specific enzymes, called galactosyltransferases, transfer galactose from the nucleotide sugar to growing sugar chains, while releasing the attached UDP. UDP is then broken down within the Golgi into UMP and transported out of the Golgi and into the cytoplasm by UGT17,19.
Disease Mechanism
Mutations in the SLC35A2 gene result in impaired UGT, restricting the amount of UDP-galactose available within the Golgi for glycosylation3. Defective UGT can restrict or prevent UDP-galactose from being transported into the Golgi, leading to glycans that lack galactose23.
Signs & Symptoms
Clinical Presentation
Individuals with SLC35A2-CDG typically develop signs and symptoms during infancy. SLC35A2-CDG is primarily characterized by global developmental delay, reductions in brain volume, alongside abnormalities in skeletal structure1,4,6,7,24–33.
- Neurological – brain region abnormalities (cerebellum and corpus collosum), abnormal brain function and brain damage (encephalopathy), abnormal brain rhythms (hypsarrythmia), intellectual disability, involuntary muscle spasms (West Syndrome), low muscle tone (hypotonia), epileptic seizures, and developmental delay
- Dysmorphic features – abnormal skull structure (brachycephaly), short nose, broad and thick eyebrows, low set ears, thick lips, coarse facial features, shortened limbs, and hand and finger abnormalities
- Ophthalmological – loss of vision (retinitis pigmentosa), involuntary rhythmic movement of eyes (nystagmus), and crossed eyes (strabismus)
- Growth problems – failure to gain weight and grow at expected rate and short stature
Other observed symptoms of SLC35A2-CDG include, but are not limited to, immune defects, spinal deformities, failure to thrive in infancy, enlarged spleen, hearing problems, and functional and structural heart defects. Other noted symptoms include curvature of spine (scoliosis), inability to walk, early onset of puberty, inverted nipples, and acid reflux.
Biochemical Abnormalities
Biochemical abnormalities observed in individuals with SLC35A2-CDG may include elevated thyroglobulin levels and liver enzymes, anemia, and abnormal coagulation factors33.
Classification
SLC35A2-CDG is classified as a disorder of multiple glycosylation pathways and more specifically, a disorder of nucleotide sugar synthesis and transport.
Diagnosis
Although diagnosis of SLC35A2-CDG may be suspected based on presentation of symptoms and a detailed patient history, genetic testing is the only definitive diagnostic test. The first-line screening tests in suspected is the analysis of serum transferrin. Measuring UDP-galactose transport activity in patient fibroblasts can also be used to identify pathogenic SLC35A2 mutations24. Although SLC35A2-CDG can also affect O-glycans, analysis of apolipoprotein C-III for O-glycan abnormalities has not been informative21,34.
Transferrin Analysis
Individuals with SLC35A2-CDG can show a type 2 pattern by transferrin isoelectric focusing (TIEF) and mass spectrometry, displaying a truncated pattern lacking galactose and terminal sialic acid. This pattern is caused by defects in the processing of the N-glycans attached to proteins within the Golgi, resulting in increased di-sialo and tri-sialotransferrin35–37.
An abnormal transferrin pattern has been observed only in 22 out of 66 SLC35A2-CDG patients reported in the literature, and this pattern has been shown to normalize with age3. As such, transferrin analysis is not a reliable method to diagnose SLC35A2-CDG37.
UDP-Galactose Transport Assay
Although not a routine diagnostic yet, an assay has recently been developed to assess transport of UDP-galactose by UGT into the Golgi in fibroblasts37.
Biomarkers
A biomarker that is unique to SLC35A2-CDG has not been identified.
Prognosis
Prognosis of SLC35A2-CDG may vary depending on the severity of an individual’s symptoms. While symptoms can be observed in infancy, most do not get diagnosed before the age of three. SLC35A2-CDG also has a low mortality rate, with only one death reported in over 60 patients characterized so far24.
Management
There are currently no treatment options approved for SLC35A2-CDG. However, replacement therapy consisting of oral D-galactose supplementation is presently being tested to treat this condition. In a clinical trial, supplementation with D-galactose to 10 patients improved various neurological and motor symptoms of SLC35A2-CDG, while increasing the levels of both fully formed sugars as well as galactosylated N-glycans8.
Research Models
Several SLC35A2 research models have been generated including yeast, worm, fly, zebrafish, and mouse models as well as patient-derived fibroblast cell lines.
Yeast (S. cerevisiae)
The yeast gms1 gene encodes a UDP galactose transporter and is orthologous to the human SLC35A2 gene. Mutations in this gene results in defective cellular glycosylation in yeast, with gene deletions resulting in the absence of galactose residues in glycoproteins found on the surface38.
Worm (C. elegans)
The C. elegans gene nstp5 encodes a transmembrane UDP-galactose transporter and is orthologous to the human SLC35A2 gene. It is a potential model for the human disease SLC35A2-CDG39.
Fly (D. melanogaster)
The Drosophila ugalt gene encodes a transporter involved UDP-galactose and galactose transmembrane transport40. It is orthologous to the human SLC35A2 gene, and mutations in this protein can destabilize and disrupt the localization of the transporter within in the fly41.
Zebrafish (D. rerio)
The slc35a2 gene in zebrafish is orthologous to the human SLC35A2 gene and could be a model for studying SLC35A2-CDG42.
Mouse (M. musculus)
Embryonic stem cell lines from mice with the Slc35a2 gene mutated have been generated43.
Human Cell Lines
SLC35A2-CDG patient-derived fibroblasts
Primary SLC35A2-CDG patient fibroblasts can be used to decipher the activity of the UDP-Galactose transporter activity in patients harbouring various mutations. In patients, loss of SLC35A2 in the fibroblasts results in reduced UDP-galactose levels in the Golgi, leading to truncated N- and O-linked glycans1.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including SLC35A2-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Groups/Organizations
SLC35A2 - CDG Type 2M Parent Support Facebook Group
Publications
SLC35A2-CDG Scientific Articles on Pubmed
Additional Resources
Heiznen lab (SLC35A2 researchers)
SLC35A2-CDG infographics
IEMbase
OMIM
Orphanet
GARD
NORD
MedlinePlus
Genetic Testing Registry
ClinVar
GeneCards
Uniprot
References
- Ng, B. G. et al. Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. American Journal of Human Genetics 92, (2013).
- Abuduxikuer, K. & Wang, J. S. Four New Cases of SLC35A2-CDG With Novel Mutations and Clinical Features. Frontiers in Genetics 12, (2021).
- Quelhas, D. et al. SLC35A2-CDG: Novel variant and review. Molecular Genetics and Metabolism Reports 26, (2021).
- Kimizu, T. et al. A case of early onset epileptic encephalopathy with de novo mutation in SLC35A2: Clinical features and treatment for epilepsy. Brain and Development 39, (2017).
- Dörre, K. et al. A new case of UDP-galactose transporter deficiency (SLC35A2-CDG): molecular basis, clinical phenotype, and therapeutic approach. Journal of Inherited Metabolic Disease 38, (2015).
- SSIEM 2016 Annual Symposium - Abstracts. Journal of Inherited Metabolic Disease 39, (2016).
- Hesse, A. N., Bevilacqua, J., Shankar, K. & Reddi, H. V. Retrospective genotype-phenotype analysis in a 305 patient cohort referred for testing of a targeted epilepsy panel. Epilepsy Research 144, (2018).
- Witters, P. et al. Clinical and biochemical improvement with galactose supplementation in SLC35A2-CDG. Genetics in Medicine 22, (2020).
- Abuduxikuer, K. & Wang, J. S. Four New Cases of SLC35A2-CDG With Novel Mutations and Clinical Features. Frontiers in Genetics 12, (2021).
- Acuna-Hidalgo, R., Veltman, J. A. & Hoischen, A. New insights into the generation and role of de novo mutations in health and disease. Genome Biology vol. 17 (2016).
- Evers, C. et al. Impact of clinical exomes in neurodevelopmental and neurometabolic disorders. Molecular Genetics and Metabolism 121, (2017).
- Foulkes, W. D. & Real, F. X. Many mosaic mutations. Current Oncology vol. 20 (2013).
- Ng, B. G. et al. Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. American Journal of Human Genetics 92, (2013).
- Song, Z. Roles of the nucleotide sugar transporters (SLC35 family) in health and disease. Molecular Aspects of Medicine vol. 34 (2013).
- Hadley, B. et al. Structure and function of nucleotide sugar transporters: Current progress. Computational and Structural Biotechnology Journal vol. 10 (2014).
- Hadley, B. et al. Nucleotide Sugar Transporter SLC35 Family Structure and Function. Computational and Structural Biotechnology Journal vol. 17 (2019).
- Sprong, H. et al. Association of the golgi UDP-galactose transporter with UDP-galactose:ceramide galactosyltransferase allows UDP-galactose import in the endoplasmic reticulum. Molecular Biology of the Cell 14, (2003).
- Ishida, N., Miura, N., Yoshioka, S. & Kawakita, M. Molecular cloning and characterization of a novel isoform of the human UDP-galactose transporter, and of related complementary DNAs belonging to the nucleotide-sugar transporter gene family. Journal of Biochemistry 120, (1996).
- Hirschberg, C. B., Robbins, P. W. & Abeijon, C. Transporters of nucleotide sugars, ATP, and nucleotide sulfate in the endoplasmic reticulum and Golgi apparatus. Annual Review of Biochemistry vol. 67 (1998).
- Varki, A. et al. Essentials of glycobiology, third edition. Cold Spring Harbor Laboratory Press (2017).
- Quelhas, D. et al. SLC35A2-CDG: Novel variant and review. Molecular Genetics and Metabolism Reports 26, (2021).
- Mikkola, S. Nucleotide sugars in chemistry and biology. Molecules vol. 25 (2020).
- Ng, B. G. et al. SLC35A2‐CDG: Functional characterization, expanded molecular, clinical, and biochemical phenotypes of 30 unreported Individuals. Human Mutation (2019) doi:10.1002/humu.23731.
- Ng, B. G. et al. SLC35A2-CDG: Functional characterization, expanded molecular, clinical, and biochemical phenotypes of 30 unreported Individuals. Human Mutation 40, (2019).
- Kodera, H. et al. De Novo Mutations in SLC35A2 Encoding a UDP-Galactose Transporter Cause Early-Onset Epileptic Encephalopathy. Human Mutation 34, (2013).
- Appenzeller, S. et al. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. American Journal of Human Genetics 95, (2014).
- Hino-Fukuyo, N. et al. Genomic analysis identifies candidate pathogenic variants in 9 of 18 patients with unexplained West syndrome. Human Genetics 134, (2015).
- Bruneel, A. et al. Complementarity of electrophoretic, mass spectrometric, and gene sequencing techniques for the diagnosis and characterization of congenital disorders of glycosylation. Electrophoresis 39, (2018).
- Westenfield, K. et al. Mosaicism of the UDP-Galactose transporter SLC35A2 in a female causing a congenital disorder of glycosylation: A case report. BMC Medical Genetics 19, (2018).
- Yates, T. M. et al. SLC35A2-related congenital disorder of glycosylation: Defining the phenotype. European Journal of Paediatric Neurology 22, (2018).
- Demos, M. et al. Diagnostic yield and treatment impact of targeted exome sequencing in early-onset epilepsy. Frontiers in Neurology 10, (2019).
- Miyamoto, S. et al. A case of de novo splice site variant in SLC35A2 showing developmental delays, spastic paraplegia, and delayed myelination. Molecular Genetics and Genomic Medicine 7, (2019).
- Vals, M. A. et al. Clinical, neuroradiological, and biochemical features of SLC35A2-CDG patients. Journal of Inherited Metabolic Disease 42, (2019).
- Wada, Y. & Okamoto, N. Apolipoprotein C-III O-glycoform profiling of 500 serum samples by matrix-assisted laser desorption/ionization mass spectrometry for diagnosis of congenital disorders of glycosylation. Journal of Mass Spectrometry 56, (2021).
- Dörre, K. et al. A new case of UDP-galactose transporter deficiency (SLC35A2-CDG): molecular basis, clinical phenotype, and therapeutic approach. Journal of Inherited Metabolic Disease 38, (2015).
- Ng, B. G. et al. Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. American Journal of Human Genetics 92, (2013).
- Ng, B. G. et al. SLC35A2‐CDG: Functional characterization, expanded molecular, clinical, and biochemical phenotypes of 30 unreported Individuals. Human Mutation (2019) doi:10.1002/humu.23731.
- Tanaka, N. & Takegawa, K. Functional characterization of Gms1p/UDP-galactose transporter in Schizosaccharomyces pombe. Yeast 18, (2001).
- Kim, W., Underwood, R. S., Greenwald, I. & Shaye, D. D. Ortholist 2: A new comparative genomic analysis of human and caenorhabditis elegans genes. Genetics 210, (2018).
- FlyBase Gene Report: Dmel\Ugalt. https://flybase.org/reports/FBgn0024994.
- Segawa, H., Kawakita, M. & Ishida, N. Human and Drosophila UDP-galactose transporters transport UDP-N-acetylgalactosamine in addition to UDP-galactose. European Journal of Biochemistry 269, (2002).
- ZFIN Gene: slc35a2. http://zfin.org/ZDB-GENE-030616-62.
- IMSR Summary. http://www.findmice.org/summary?gaccid=MGI:1345297.