
Lay Summary
POMT1-CDG is a rare inherited condition that affects many parts of the body. To date, there are at least 105 patients reported in the medical literature. POMT1-CDG is classified as a disorder of O-linked protein glycosylation and more specifically, a disorder of O-mannosylation. POMT1-CDG is also classified as a dystroglycanopathy, which refers to a group disorder that affects the glycosylation of the protein alpha-dystroglycan. The alpha-dystroglycan protein plays an important role in muscle, brain and eye development. POMT1-CDG is caused when an individual has mutations in both copies of their POMT1 gene, which encodes an enzyme that attaches the simple sugar mannose to a protein during O-glycosylation. Mutations in the POMT1 gene cause proteins to be under glycosylated, meaning these proteins have less sugars attached than healthy forms of the proteins. POMT1-CDG can present as three different forms that range in severity, with symptoms beginning from birth or in adolescence depending on the form. POMT1-CDG is primarily characterized by muscle, brain, eye, and heart problems. Screening tests are available for POMT1-CDG, but a definitive diagnosis is achieved through genetic testing. Treatment is focused on the management of specific symptoms and preventing complications.
Overview
Protein O-mannosyltransferase 1 congenital disorder of glycosylation (POMT1-CDG) is a rare autosomal recessive genetic disorder. The first case of POMT1-CDG was reported in 20021, and there are at least 105 reported cases in the literature to date1–18. The POMT1 gene encodes an enzyme, protein O-mannosyltransferase 1 (POMT1), which is responsible for adding the first mannose to serine or threonine residues on a protein during O-glycosylation. Deficiency of POMT1 results in deficient O-mannosylation of the the protein alpha-dystroglycan—a protein that is part of the dystrophin-glycoprotein complex that is needed for a range of roles, such as stabilizing muscle tissue and cell signalling19-20. POMT1-CDG is also classified as a dystroglycanopathy, which refers to a group disorders that affect O-mannosylation of alpha-dystroglycan13.
POMT1-CDG may present as three different forms; Muscular dystrophy-dystroglycanopathy -type A1, -type B1 or -type C1, which vary in clinical presentation and symptom severity. The characteristic clinical presentation of POMT1-CDG includes muscle, brain, and eye problems. Definitive diagnosis can only be achieved through molecular genetic testing. There are currently no approved treatments for POMT1-CDG.
Synonyms
- Autosomal recessive limb-girdle muscular dystrophy type 2K
- Limb-girdle muscular dystrophy type 2K (LGMD2K)
- Limb-girdle muscular dystrophy-intellectual disability syndrome
- POMT1-related LGMD R11
- POMT1-related hydrocephalus
- Muscular dystrophy-dystroglycanopathy, type A1 (MDDGA1)
- Muscular dystrophy-dystroglycanopathy, type B1 (MDDGB1)
- Muscular dystrophy-dystroglycanopathy, type C1 (MDDGC1)
Inheritance
POMT1-CDG is inherited in an autosomal recessive fashion, where a mutation is inherited from each parent (asymptomatic carriers).
Gene Function
POMT1 encodes a mannosyltransferase enzyme, protein O-mannosyltransferase 1 (POMT1). Mannosyltransferases are enzymes that enable the transfer of mannose during glycosylation. POMT1 is located in the ER membrane, with exposed sections in the cytoplasm and the ER lumen. POMT1 adds mannose to a serine or threonine amino acid residue of a protein during O-glycosylation in the ER13. POMT1 must form a complex with a separate enzyme, O-mannosyltransferase 2 (POMT2), to transfer mannose to a protein21. Alpha-dystroglycan is an O-mannosylated protein that has been reported to be abnormally glycosylated in POMT1-CDG patients.
O-mannosylation of alpha-dystroglycan
Alpha-dystroglycan is an O-mannosylated glycoprotein that is found on various cells, such as muscle, heart, and nerve22. O-mannosylation is a type of O-glycosylation which involved the attachment of mannose to serine or threonine residues on a protein. Alpha-dystroglycan is the central component of the dystrophin-glycoprotein complex, which links the extracellular matrix to the intracellular cytoskeleton of muscle cells. Glycosylated alpha-dystroglycan binds to components of the extracellular matrix, such as laminin, and to beta-dystroglycan—which is a protein that is in the cell membrane that joins to the intracellular protein dystrophin23. Alpha-dystroglycan needs to be O-glycosylated to bind to laminin. Alpha-dystroglycan is also needed for a variety of other processes, such as development and cell signalling19,23.
Alpha-dystroglycan O-glycans can have variable structures, where different tissues can have alpha-dystroglycan with different O-glycans. The o-mannosylated glycans on alpha-dystroglycan can be categorized into three core structures – M1, M2, and M3 – which can then be further modified (Figure 1) 24.
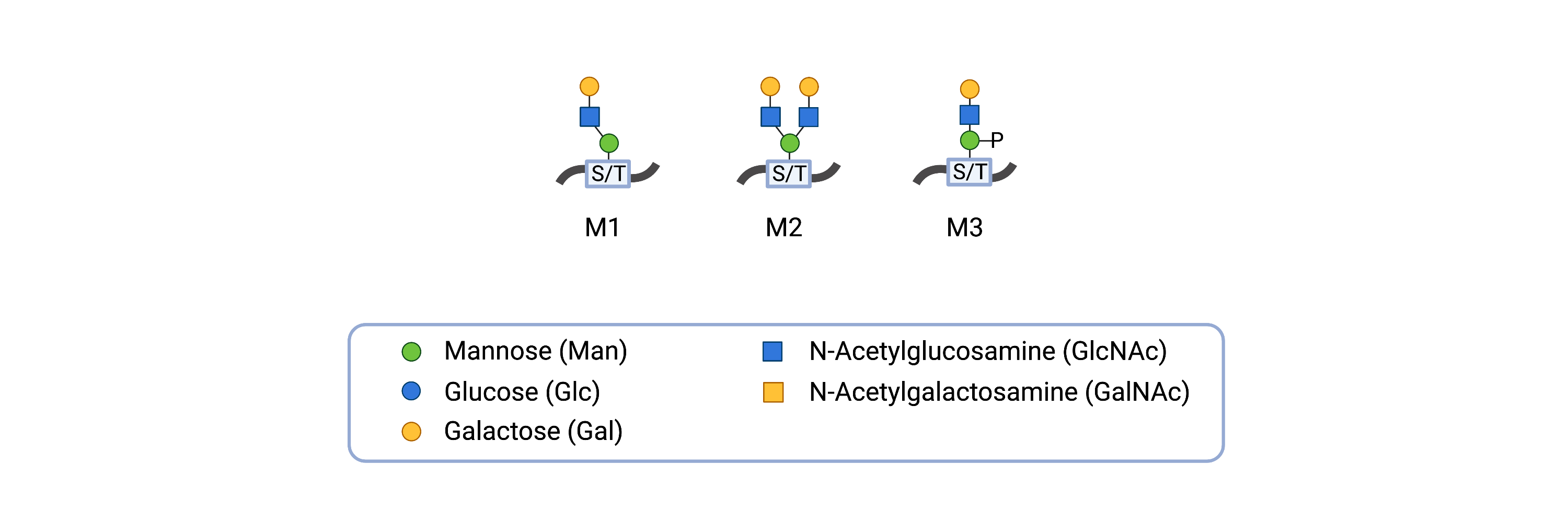
Figure 1. O-mannosyl glycan core structures of alpha-dystroglycan.
The o-mannosylated glycans on alpha-dystroglycan can be categorized as cores M1, M2, or M3 and can be extended by the attachment of additional monosaccharides to the glycan.
However, the first step of O-mannosylation of alpha-dystroglycan requires an initial mannose to be added to the protein. For this, POMT1 forms a complex with POMT2 and transfers mannose from dolichol-phosphate mannose (Dol-P-mannose) to a serine or threonine residue on alpha-dystroglycan in the ER21. Depending on the final structure, additional enzymes modify the O-glycan in the ER and/or Golgi (Figure 2)25.
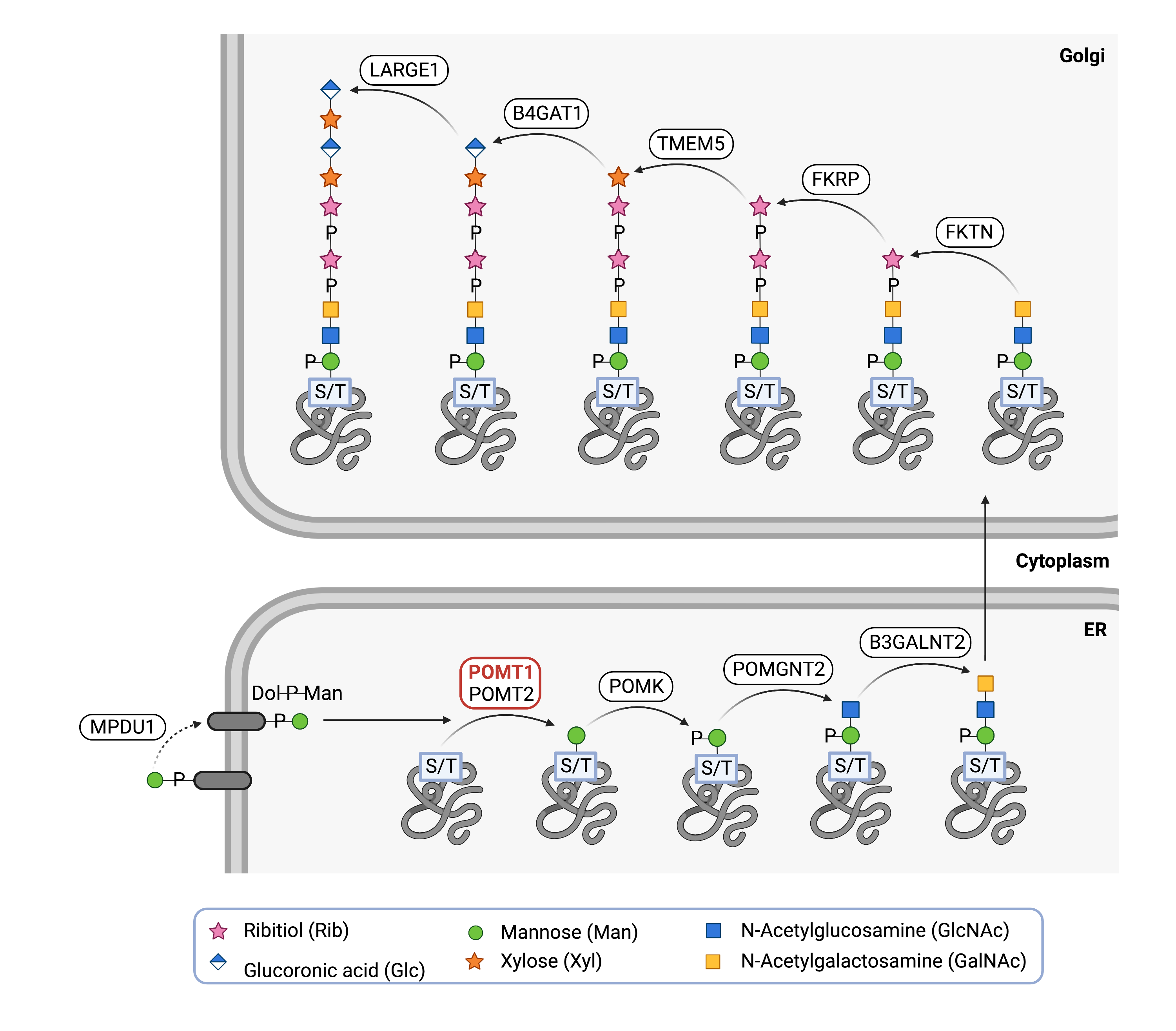
Figure 2.Role of POMT1 in o-mannosylation of alpha-dystroglycan.
POMT1, along with POMT2, adds the first mannose in the glycan chain to a serine or threonine residue of a protein, such as alpha-dystroglycan. Additional enzymes then add additional monosaccharides to generate the glycan chain on alpha-dystroglycan.
Disease Mechanism
Mutations in the POMT1 gene lead to POMT1 having a reduced ability or inability to transfer mannose onto a protein during O-glycosylation. Alpha-dystroglycan has been reported to be hypoglycosylated in POMT1-CDG patients. As alpha-dystroglycan is present in various tissues, such as muscle, nerve, and eye, clinical features of POMT1-CDG may be due to hypoglycosylation of alpha-dystroglycan, which prevents alpha-dystroglycan from functioning properly26,27.
Mutations
The POMT1 gene is located on chromosome 9 (9q34.13). At least 76 pathogenic mutations have been reported for the POMT1 gene, such as missense, deletion, splicing, nonsense, and insertion mutations20. The various phenotypes of POMT1-CDG are linked to the level of POMT1 activity. More severe phenotypes, like Walker-Warburg syndrome, are caused my mutations resulting in stop codons or deletions in highly conserved domains, resulting in truncated proteins and disrupting POMT1 activity27. A milder phenotype is found in patients with missense mutations. The mutation, p.Ala200Pro, has been found in patients who present with no brain malformations but with intellectual disability 5.
Signs & Symptoms
Clinical Presentation
Individuals with POMT1-CDG can be classified into three phenotypes which range in their severity: Muscular dystrophy-dystroglycanopathy type A1, type B1 and type C1. 20,27:
Muscular dystrophy-dystroglycanopathy type A1 (MDDGA1)
MDDGA1 is a severe form of congenital muscular dystrophy-dystroglycanopathy, typically presenting prenatally or at birth and is characterized by brain and eye defects. It includes disorders such as Walker-Warburg syndrome (WWS) and muscle-eye-brain disease (MEB). The clinical presentation may include a combination of:
- Neurological – a build-up of fluid in the brain (hydrocephalus), defective brain development, a sac-like bulge that pokes out of the skull (encephalocele), intellectual disability, and seizures
- Musculoskeletal – hypotonia, delayed milestones, contractures, and impaired ability to walk and speech
- Ophthalmological – microphthalmia, exophthalmia, enlarged eye, megalocornea, glaucoma, hypertelorism, and myopia
Muscular dystrophy-dystroglycanopathy type B1 (MDDGB1)
MDDGB1 is a moderate severe form of congenital muscular dystrophy-dystroglycanopathy with or without intellectual disability. MDDGB1 clinical presentation is milder than MDDGA1 and may include a combination of:
- Neurological – intellectual disability, microcephaly, and cerebellar hypoplasia
- Musculoskeletal – hypotonia, contractures, delayed motor milestones, scoliosis, and hypertrophy
- Ophthalmological – myopia, retinal degeneration, and cataracts
Muscular dystrophy-dystroglycanopathy type C1 (MDDGC1)
MDDGC1 is a mild form of limb-girdle muscular dystrophy with or without intellectual disability, presenting with late-onset progressive muscle weakness. MDDGC1 clinical presentation may include a combination of:
- Neurological – intellectual disability
- Musculoskeletal – delayed motor milestones, hypertrophy, limb–girdle weakness, muscle weakness, dystrophic and myopathic changes
- Cardiac – cardiomyopathy
Other clinical presentations of POMT1-CDG may involve cardiac defects, absence of speech and unable to walk1–18.
Biochemical Abnormalities
Biochemical abnormalities observed in individuals with POMT1-CDG include elevated serum creatine levels.
Classification
POMT1-CDG is classified as a disorder of O-linked protein glycosylation and, within this group, a disorder of O-mannosylation.
Diagnosis
Although diagnosis of POMT1-CDG may be suspected based on the presentation of symptoms and a detailed patient history, direct molecular genetic testing is the only definitive diagnostic test. Screening tests in suspected patients can involve immunostaining of alpha-dystroglycan from muscle cell samples and POMT1 enzyme analysis.
Alpha-dystroglycan staining
Muscle biopsies taken from patients are stained or immunolabeled with antibodies that bind to glycosylated alpha-dystroglycan. Consequently, hypoglycosylated alpha-dystroglycan will have lower binding levels and immunolabeling than healthy alpha-dystroglycan. More severe POMT1-CDG phenotypes have less immunolabelling that milder forms28.
POMT1 Enzyme Analysis
POMT1 enzyme activity is measured using patient fibroblasts samples. POMT1 activity is reduced in POMT1-CDG samples compared to control samples, with activity being more impaired with more severe POMT1-CDG phenotypes10.
Biomarkers
There are currently no biomarkers described for POMT1-CDG.
Prognosis
Prognosis of POMT1-CDG may vary depending on the severity of an individual’s symptoms and the specific form. Severely affected patients, such as those with MDDGA1, may not survive beyond the first few months of life, with some surviving to the age of 327,29. Less severely affected patients, such as those with MDDGC1, may not present symptoms until adulthood12,27.
In patients with less severe types of POMT1-CDG, patients usually achieve mobility before the onset of progressive muscular weakness. The progressive muscle weakness typically first affects voluntary muscles in the shoulder and pelvic gridles27.
Management
Management of symptoms may include combinations of physical therapy, occupational therapy, vision therapy, and palliative measures.
Therapies
There are currently no treatment options available for POMT1-CDG. Treatment is focused on the management of symptoms and prevention of complications.
Research Models
Several POMT1 models have been generated, such as in flies, fish, mice, and human cells.
Fly (D. melanogaster)
Drosophila gene dPOMT1 is orthologous to human POMT1. Mutations in dPOMT1 can lead to synaptic defects30. Glycosylation of Dg by POMT1 and POMT2 are important to Dg function in maintaining cell integrity in the muscles of larval Drosophila. Mutations in dPMOT1 in larvae show defects in muscle attachment and contraction in phenotypes with impaired or hypoglycosylated dystroglycan31.
Fish (D. rerio)
POMT1 orthologue has been identified in zebrafish32.
Mouse (M. musculus)
Pomt1 Expression in Mice
Mouse gene mPomt1 is orthologous to human POMT1 and expression is detected in different mouse tissues, such as muscle, eye, brain, and testes33.
Homozygous Pomt1-/- Knockout Mice
Pomt1 knockout mice were found to be embryonic lethal between embryonic days 7.5 and 9.534.
Pomt1 Photoreceptor-specific Conditional Knockout Mice
Knockout of Pomt1 in the photoreceptor cells in mice showed impaired alpha-dystroglycan O-mannosylation as well as additional effects, such as morphological changes35.
Human Cell Lines
Human Embryonic Kidney Stem Cells (HEK293T)
Plasmids with POMT1 and POMT2 transfected into HEK293T cells showed that POMT1 and POMT2 must be co-expressed to form an active enzyme complex36.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including POMT1-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDGs, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Publications
POMT1-CDG Scientific Articles on PubMed
Additional References
OMIM – MDDGA1
OMIM – MDDGB1
OMIM – MDDGC1
IEMbase
MedlinePlus
OrphaNet
Genetic Testing Registry
ClinVar
NIH
GeneCards
UniProt
Marrvel
References
- de Bernabé, D. B. V. et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. American Journal of Human Genetics 71, (2002).
- van Reeuwijk, J. et al. The expanding phenotype of POMT1 mutations: From Walker-Warburg syndrome to congenital muscular dystrophy, microcephaly, and mental retardation. Human Mutation 27, (2006).
- Currier, S. C. et al. Mutations in POMT1 are found in a minority of patients with walker-warburg syndrome. American Journal of Medical Genetics 133 A, (2005).
- Balci, B. et al. An autosomal recessive limb girdle muscular dystrophy (LGMD2) with mild mental retardation is allelic to Walker-Warburg syndrome (WWS) caused by a mutation in the POMT1 gene. Neuromuscular Disorders 15, (2005).
- Clement, E. et al. Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Annals of Neurology 64, (2008).
- Mercuri, E. et al. Congenital muscular dystrophies with defective glycosylation of dystroglycan: A population study. Neurology 72, (2009).
- Lommel, M. et al. Correlation of enzyme activity and clinical phenotype in POMT1-associated dystroglycanopathies. Neurology 74, (2010).
- Endo, T., Manya, H., Seta, N. & Guicheney, P. POMGnT1, POMT1, and POMT2 mutations in congenital muscular dystrophies. in Methods in Enzymology vol. 479 (2010).
- Song, D. et al. Genetic variations and clinical spectrum of dystroglycanopathy in a large cohort of Chinese patients. Clinical Genetics 99, (2021).
- Yang, H. et al. Analysis of phenotype, enzyme activity and genotype of Chinese patients with POMT1 mutation. Journal of Human Genetics 61, (2016).
- Hu, P. et al. Compound heterozygous POMT1 mutations in a Chinese family with autosomal recessive muscular dystrophy-dystroglycanopathy C1. Journal of Cellular and Molecular Medicine 21, (2017).
- Bello, L. et al. Cardiomyopathy in patients with POMT1-related congenital and limb-girdle muscular dystrophy. European Journal of Human Genetics 20, (2012).
- Geis, T. et al. Clinical long-time course, novel mutations and genotype-phenotype correlation in a cohort of 27 families with POMT1-related disorders. Orphanet Journal of Rare Diseases 14, (2019).
- Chong, Y. K. et al. Dystroglycanopathy with two novel POMT1 mutations in a Chinese boy with developmental delay and muscular dystrophy. European Journal of Paediatric Neurology 18, (2014).
- Pane, M. et al. Respiratory and cardiac function in congenital muscular dystrophies with alpha dystroglycan deficiency. Neuromuscular Disorders 22, (2012).
- Yis, U. et al. A case of Walker-Warburg syndrome resulting from a homozygous POMT1 mutation. European Journal of Paediatric Neurology 11, (2007).
- Judaš, M. et al. POMT1-associated walker-warburg syndrome: A disorder of dendritic development of neocortical neurons. Neuropediatrics 40, (2009).
- Gan, S., Yang, H., Xiao, T., Pan, Z. & Wu, L. POMT1 and POMT2 gene mutations result in 2 cases of alpha-dystroglycanopathy. Zhong Nan Da Xue Xue Bao Yi Xue Ban 46, (2021).
- Balci-Hayta, B., Talim, B., Kale, G. & Dincer, P. LARGE expression in different types of muscular dystrophies other than dystroglycanopathy. BMC Neurology 18, (2018).
- Hu, P., Yuan, L. & Deng, H. Molecular genetics of the POMT1-related muscular dystrophy-dystroglycanopathies. Mutation Research - Reviews in Mutation Research vol. 778 (2018).
- Akasaka-Manya, K., Manya, H., Nakajima, A., Kawakita, M. & Endo, T. Physical and functional association of human protein O-mannosyltransferases 1 and 2. Journal of Biological Chemistry 281, (2006).
- Hewitt, J. E. Abnormal glycosylation of dystroglycan in human genetic disease. Biochimica et Biophysica Acta - Molecular Basis of Disease vol. 1792 (2009).
- Endo, T. Glycobiology of alpha-dystroglycan and muscular dystrophy. Journal of Biochemistry 157, (2015).
- Yoshida-Moriguchi, T. & Campbell, K. P. Matriglycan: A novel polysaccharide that links dystroglycan to the basement membrane. Glycobiology vol. 25 (2014).
- Praissman, J. L. et al. The functional O-mannose glycan on α-dystroglycan contains a phospho-ribitol primed for matriglycan addition. Elife 5, (2016).
- Godfrey, C. et al. Refining genotype–phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 130, 2725–2735 (2007).
- Hu, P., Yuan, L. & Deng, H. Molecular genetics of the POMT1-related muscular dystrophy-dystroglycanopathies. Mutation Research/Reviews in Mutation Research 778, 45–50 (2018).
- Jimenez-Mallebrera, C. et al. A comparative study of α-dystroglycan glycosylation in dystroglycanopathies suggests that the hypoglycosylation of α-dystroglycan does not consistently correlate with clinical severity. Brain Pathology 19, (2009).
- Kim, D. S. et al. POMT1 mutation results in defective glycosylation and loss of laminin-binding activity in α-DG. Neurology 62, (2004).
- Wairkar, Y. P., Fradkin, L. G., Noordermeer, J. N. & DiAntonio, A. Synaptic defects in a Drosophila model of congenital muscular dystrophy. Journal of Neuroscience 28, (2008).
- Haines, N., Seabrooke, S. & Stewart, B. A. Dystroglycan and protein O-mannosyltransferases 1 and 2 are required to maintain integrity of Drosophila larval muscles. Molecular Biology of the Cell 18, (2007).
- Moore, C. J., Goh, H. T. & Hewitt, J. E. Genes required for functional glycosylation of dystroglycan are conserved in zebrafish. Genomics 92, (2008).
- Prados, B., Peña, A., Cotarelo, R. P., Valero, M. C. & Cruces, J. Expression of the murine Pomt1 gene in both the developing brain and adult muscle tissues and its relationship with clinical aspects of Walker-Warburg syndrome. American Journal of Pathology 170, (2007).
- Willer, T. et al. Targeted disruption of the Walker-Warburg syndrome gene Pomt1 in mouse results in embryonic lethality. Proc Natl Acad Sci U S A 101, (2004).
- Uribe, M. L. et al. Retinal Proteomics of a Mouse Model of Dystroglycanopathies Reveals Molecular Alterations in Photoreceptors. Journal of Proteome Research 20, (2021).
- Manya, H. et al. Demonstration of mammalian protein O-mannosyltransferase activity: Coexpression of POMT1 and POMT2 required for enzymatic activity. Proc Natl Acad Sci U S A 101, (2004).