
Lay Summary
PIGW-CDG, also known as hyperphosphatasia with mental retardation syndrome type 5 (HPMRS5) or Mabry syndrome, is a rare inherited condition that affects multiple systems in the body. To date, fewer than 20 cases have been reported in the medical literature. PIGW-CDG is classified as a disorder of GPI-anchor biosynthesis and is caused by mutations in both copies of the PIGW gene. This gene provides instructions for making a protein that helps build GPI anchors which are molecules that attach certain proteins to the cell surface. The PIGW protein attaches a fatty acid to the GPI anchor, a step that is essential for attaching GPI-anchored proteins to the cell surface. Mutations in the PIGW gene cause defects in GPI-anchored proteins, making them unstable or unable to attach to the cell surface. Symptoms of PIGW-CDG typically appear in infancy and vary widely among affected individuals. Symptoms are mainly characterized by epilepsy, developmental delay, intellectual disability, low muscle tone, elevated levels of serum alkaline phosphatase (hyperphosphatasia), distinctive features and various congenital anomalies. Other reported symptoms include recurrent respiratory infections, hernia in the groin, hearing problems, nystagmus, brain and cardiac anomalies. Some individuals diagnosed with PIGW-CDG may have a lower life expectancy due to complications arising from symptom complications and congenital anomalies. PIGW-CDG is usually diagnosed through genetic testing, often combined with biochemical tests that measure proteins released into the blood stream under certain conditions of GPI deficiency. Additionally, testing for the presence of GPI–anchored proteins on certain blood cells can also help screen for PIGW-CDG. There are currently no approved treatments for PIGW–CDG. Current medical care is focused on managing symptoms and preventing complications.
Overview
Phosphatidylinositol glycan class W congenital disorder of glycosylation (PIGW-CDG), also commonly referred to as hyperphosphatasia with mental retardation syndrome type 5 (HPMRS5) or Mabry syndrome, is a rare autosomal recessive genetic disorder. The first reported case of PIGW–CDG was in 2014, initially diagnosed as having West syndrome, a severe form of infantile epilepsy1. As of 2025, 16 cases have been reported in the literature, including 4 fetuses1–9. The autosomal recessive condition is caused by biallelic pathogenic variants of the PIGW gene1,5. However, in one reported case, the patient carried a heterozygous variant in PIGW and a deletion of chromosome 17q12 which includes the PIGW gene6.
The PIGW gene encodes an enzyme that catalyzes the attachment of a fatty acid to the GPI intermediate, glucosamine phosphatidylinositol (GlcN-PI), during GPI–anchored protein biosynthesis10. GPI anchors are an important mechanism of attachment for proteins to the cell surface, and these proteins are known as GPI-anchored proteins (GPI-APs). At least 150 proteins are GPI-APs11. Mutations in the PIGW gene result in defects in GPI-APs, which are critical to the development and function of many organs, especially the neurological, ophthalmological, musculoskeletal, gastrointestinal, respiratory, and skeletal systems1–9.
Clinical manifestations typically arise in the newborn period or early infancy, but the severity and presentation can vary widely. Common symptoms include early-onset epileptic seizures, developmental delay, intellectual disability, hypotonia, elevated levels of serum alkaline phosphatase (hyperphosphatasia), dysmorphic features and various congenital anomalies1–9. Additional symptoms such as recurrent respiratory infections, cardiovascular defects, inguinal hernia, auditory pathway defects and nystagmus have also been reported6,8.
PIGW-CDG is not included in newborn screening panels and is typically diagnosed through whole-exome or whole-genome sequencing, often combined with biochemical tests, such as serum alkaline phosphatase levels and flow cytometry to assess GPI-anchored protein expression on patient cells 1,2,6,8.
Due to its rarity, the natural history and long-term prognosis of PIGW-CDG are unknown. Treatment is focused on managing symptoms while preventing complications. Epileptic seizures are often severe and refractory to anti-seizure medications4,6,8.
Synonyms
- PIGW Deficiency
- Hypophosphatasia with mental retardation syndrome 5 (HPMRS5)
- Glycosylphosphatidylinositol biosynthesis defect 11 (GPIBD11)
- Congenital Disorder of Glycosylation due to PIGW deficiency
- Mabry Syndrome
Inheritance
PIGW-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The PIGW gene encodes an enzyme (PIG-W) that is involved in the early steps of GPI-anchor biosynthesis. PIG-W is an acyltransferase enzyme that attaches a fatty acid (acyl chain) to inositol on the GPI intermediate glucosamine phosphatidylinositol (GlcN-PI). This process, known as acylation (specifically inositol acylation), occurs in the endoplasmic reticulum (ER) and involves transferring a fatty acid from an acyl-CoA molecule (a type of activated fatty acid donor) to the inositol ring of GlcN-PI. In humans, the acyl-CoA donor is typically palmitoyl-CoA (a 16-carbon saturated fatty acyl-CoA derived from palmitic acid), although PIG-W can also transfer acyl chains of varying lengths10.
The attachment of a fatty acid to the GPI intermediate by PIG-W is essential because it ensures the proper assembly of the GPI anchor, allowing it to attach correctly to proteins and undergo necessary remodeling steps. Later in the GPI biosynthesis pathway, the fatty acid is removed in a remodeling step, which is necessary to produce fully functional GPI-anchored proteins that attach to the cell surface10–12.
GPI-ANCHORED PROTEIN BIOSYNTHESIS
GPI-anchored protein biosynthesis is one of the major glycosylation pathways that attach glycans to lipid molecules within cells. Many proteins, called GPI-anchored proteins, are attached to the cell surface by GPI anchors.
The mature GPI core structure consists of phosphatidylinositol (PI), glucosamine (GlcN), three mannose sugars (Man3), and two phosphoethanolamines (EtN-P2) (Figure 1). GPI-anchored proteins are attached to the GPI by forming a bond between the EtN-P group of the GPI core and the C-terminus of newly synthesized proteins in the ER lumen. In some GPI-APs, the core glycan is modified with a fourth mannose and/or N-acetylglucosamine (Glc-NAc) side chains12.
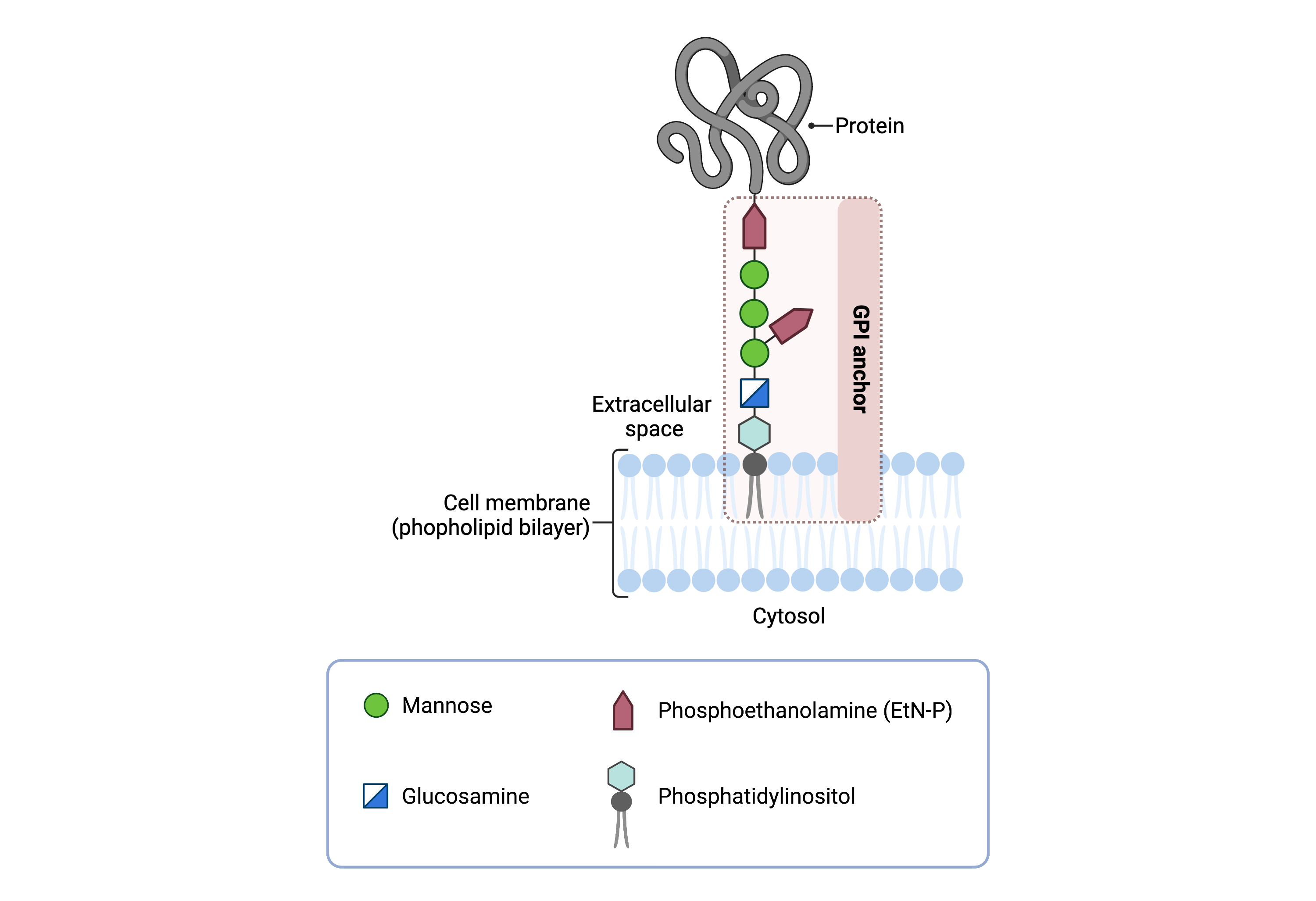
Figure 1: GPI-anchored protein structure.
The GPI anchor is a glycolipid that attaches proteins to the plasma membrane. The mammalian GPI core consists of an EtNP-Man-3-Man-2-Man-1-GlcN-PI backbone, with an EtN-P side branch linked to Man-1. The phosphatidylinositol lipid tail is embedded in the plasma membrane, where it associates with lipid rafts, facilitating protein localization and function.
The generation of a GPI-anchored proteins is a multi-step process involving more than 30 enzymes and can be divided into the following steps (Figure 2)11,13,14:
- GPI anchor synthesis
- Protein attachment
- Lipid/glycan remodelling and protein transport
GPI anchor synthesis and protein attachment is largely carried out by a series of enzymes encoded by the PIG genes, while enzymes encoded by the PGAP genes facilitate remodelling of the GPI anchored protein.

Figure 2: Overview of GPI-anchored protein biosynthesis and PIG-W function.
GPI-anchored protein biosynthesis involves a series of enzymatic reactions. Beginning on the cytoplasmic side of the ER, the GPI anchor is synthesized in a stepwise process and then attached to target proteins. After protein attachment, the glycan and lipid components of the GPI anchor undergo further modifications in the ER and Golgi, before the mature GPI-AP is transported to the cell surface. PIGW catalyzes the transfer of an acyl chain from acyl-CoA to the inositol ring of the intermediate GlcN-PI, to generate GlcN-(acyl)-PI.
GPI Anchor Synthesis
The first stage of GPI anchor biosynthesis involves the stepwise construction of the GPI anchor in the ER. This process begins on the cytoplasmic side of the ER, where N-acetylglucosamine (GlcNAc) is transferred to the lipid phosphatidylinositol (PI), by the GPI GlcNAc transferase (GPI-GnT) complex, forming GlcNAc-PI12,15. This intermediate is de-N-acetylated by PIG-L, converting it to glucosaminyl-PI (GlcN-PI) which is subsequently flipped into the lumen of the ER11,16.
Once in the ER lumen, PIG-W transfers a fatty acid from an acyl-CoA donor to the inositol ring of GlcN-PI, generating GlcN-(acyl)PI. This inositol acylation by PIG-W is a critical modification that facilitates proper GPI anchor attachment to proteins and subsequent remodeling steps necessary for GPI maturation. This modification enhances the efficiency of the attachment of the first mannose residue and may be required for the attachment of EtN-P to the third mannose by the PIG-O and PIG-F complex 10,17,18 .
As biosynthesis progresses, additional enzymes modify the GPI anchor, generating the GPI anchor core (PI-GlcN-Man3-EtN-P3)13,19.
Protein Attachment
Once the GPI anchor has been synthesized, it is transferred en bloc to a protein with a C-terminal GPI attachment signal sequence by GPI transamidase (GPI-TA). The five-protein enzyme complex catalyzes the simultaneous cleavage of the signal sequence and attachment of the GPI anchor to the newly synthesized protein11.
LIPID/GLYCAN REMODELLING AND PROTEIN TRANSPORT
After the protein has been attached to the GPI anchor, both the glycan and lipid portion of the anchor undergo modifications (referred to as remodeling) in the ER and Golgi by post-GPI attachment to protein (PGAP) enzymes or the glycosyltransferase B3GALT4. In the ER, PGAP1 removes the acyl chain previously attached by PIG-W. Following further remodelling, the mature GPI-anchored protein is then transported to the plasma membrane where it associates with other GPI-anchored proteins in lipid rafts20.
Disease Mechanism
Mutations in the PIGW gene compromise the function of the PIG-W protein, leading to partial or complete loss of enzymatic activity, consistent with a hypomorphic or loss-of-function pathogenic mechanism2,8. As a result, intermediates lacking the inositol-linked acyl chain accumulate, leading to premature activation of the GPI transamidase (GPI-TA) complex which catalyzes the attachment of GPI anchors to proteins1. This results in incomplete GPI anchors being attached to proteins, preventing their proper anchoring into the cell membrane. These proteins are either released as soluble forms, or degraded intracellularly (Figure 3)12. The loss of GPI-APs from the cell surface contributes to the pathological features of PIGW-CDG, including severe neurological phenotypes and hyperphosphatasia21,22.

Figure 3: PIGW-CDG disease mechanism.
In PIGW-CDG, intermediates lacking the inositol-linked acyl chain accumulate during GPI biosynthesis, leading to premature activation of the GPI transamidase (GPI-TA) complex and incomplete GPI anchors being attached to proteins. These proteins are either released as soluble forms, or degraded intracellularly. Hyperphosphatasia results from the release of a soluble form of alkaline phosphatase into the blood stream.
Hyperphosphatasia in PIGW-CDG results from the release of a soluble form of the GPI-anchor protein, alkaline phosphatase (ALP), into the bloodstream. The mechanisms of hyperphosphatasia vary depending on the specific gene affected and the stage of the GPI biosynthesis pathway that is disrupted. It has been proposed that mutations in genes affecting early steps of GPI biosynthesis lead to a greater degradation of ALP, whereas mutations in genes involved in later steps result in increased secretion of ALP22.
Studies have demonstrated that ALP secretion requires the GPI transamidase complex, which recognizes incomplete GPI intermediates and cleaves a hydrophobic peptide signal, causing the release of ALP into the blood stream22. Additionally, hyperphosphatasia may contribute to seizures by reducing the availability of pyridoxal 5′-phosphate (PLP), a coenzyme required for γ-aminobutyric acid (GABA) synthesis. Increased dephosphorylation of PLP by excessive ALP could lead to impaired GABA production, resulting in neuronal hyperexcitability and increased seizure susceptibility23.
Mutations
The PIGW gene is located on chromosome 17 (17q12). To date, twelve missense, one frameshift and one gene deletion in PIGW have been reported. Most of the variants fall within the transmembrane domain of PIG-W (Figure 4). Homozygous p.(Arg36Gly) has been reported in three fetuses from two unrelated families presenting with multiple congenital anomalies8. The absence of loss-of-function PIG-W variants in a compound heterozygous or homozygous state with complete GPI deficiency suggests that some residual enzymatic activity is essential for survival8. Establishing genotype-phenotype correlations in PIGW-related disorders is challenging, as both the specific mutations (e.g., missense, frameshift) and their zygosity significantly influence clinical manifestations8.

Figure 4: Schematic diagram of the PIG-W protein structure and reported pathogenic variants.
PIG-W contains 13 transmembrane domains (numbered), with loops facing either the ER lumen or the cytoplasm. Reported patient variants and their position are indicated.
Signs & Symptoms
Clinical Presentation
Individuals with PIGW-CDG typically develop signs and symptoms at birth or in early infancy. However, most reported patients had prenatal signs, often multiple congenital anomalies, suggesting that it could present early in pregnancy8. PIGW-CDG is primarily characterized by global developmental delay, seizures, hypotonia, hyperphosphatasia, congenital anomalies and dysmorphic features. Reported symptoms of PIGW-CDG include1–9:
- Neurological symptoms– global developmental delay, intellectual disability, low muscle tone (hypotonia) and a loss of muscle control (ataxia), epileptic seizures
- Dysmorphic features – facial bone abnormalities (broad nasal bridge, tented upper lip, anteverted nares), mild flexion contractures of all fingers, large tongue, structural deformity of the anterior thoracic wall (pectus excavatum)
- Ophthalmological symptoms – involuntary side-to-side movement of eyes (nystagmus), crossed eyes (strabismus), and vision problems originating from the brain (cortical visual impairment)
- Congenital anomalies – hernias (umbilical, bilateral indirect, inguinal, diaphragmatic), swollen kidney(s) due to urine build up (hydronephrosis), external reproductive organs being underdeveloped (genital hypoplasia)
- Respiratory symptoms – recurrent respiratory infections
Other symptoms include West syndrome, a type of severe epilepsy that affects infants. It is characterized by epileptic spasms, hypsarrhythmia (a highly irregular pattern of brain waves on EEG), and developmental arrest or regression. Less common symptoms of include behavioral abnormalities, cardiac abnormalities/defects (ie. bradycardia), and gastrointestinal dysfunction.
Biochemical Presentation
Biochemical abnormalities observed in individuals with PIGW-CDG include elevated alkaline phosphatase (ALP) levels. Alkaline phosphatase is a GPI-anchor protein that is normally found on the cell surface but may be secreted as a soluble protein in GPI deficiencies. Reduced or absent expression of CD16 and CD24, GPI- anchored proteins found on the surface of blood have also been reported in patients11.
Classification
PIGW-CDG is classified as a disorder of GPI anchor biosynthesis.
Diagnosis
GPI-related CDG should be considered in individuals presenting with early onset severe seizure disorders, dysmorphic facial features and hyperphosphatasia. As currently available screening tests for CDG, such as transferrin or glycan analysis, will not detect PIGW-CDG, diagnosis is typically achieved through genetic testing, either as part of an epilepsy panel or whole exome sequencing. PIGW-CDG may also be screened for by analyzing surface GPI-anchored protein expression on blood cells by flow cytometry.
Flow Cytometry
Individuals with PIGW-CDG lack GPI-anchored proteins on the surface of their granulocytes, lymphocytes, and monocytes, which are types of white blood cells25. Specifically, these GPI-anchored proteins constitute of reduced or absent expression of CD16 and CD24 from granulocytes, CD14 from monocytes, and CD59 from lymphocytes8.
Biomarkers
Elevated serum alkaline phosphatase levels have been reported in most PIGW-CDG patients, and may serve as a potential biomarker for diagnosis, disease monitoring, and assessing treatment response in PIGW-CDG8.
Prognosis
Prognosis of PIGW-CDG may vary depending on severity of an individual’s symptoms. Due to the rarity of PIGW-CDG, its natural history and long-term prognosis are currently unknown. The risk of death in cases of severe respiratory infection is present in patients with PIG-W dysfunction8. One reported patient passed away at the age 3 due to cardiac arrest related to pneumonia8. The broad clinical spectrum, based on general GPI disorder presentation, may result in severe neurological symptoms, congenital anomalies and can be life-threatening.
Management
Management of symptoms may include combinations of physical therapy, occupational therapy, and palliative measures. The majority of reported PIGW-CDG pediatric patients have early onset-epilepsy. In some patients, seizures are treatable with anti-epileptic medications, although many are often treatment-resistant8.
Therapies
There are currently no direct treatment options available for PIGW-CDG. However, Perlara PBC has discovered potential drug repurposing candidates for PIGW-CDG which are currently being evaluated in observational patient studies (read more here).
Research Models
Research models available to study PIGW-CDG currently includes a yeast model. Other model organisms may be suitable for PIGW-CDG research.
Yeast (S. cerevisiae)
GWT1 is the ortholog of the mammalian PIGW gene (SGD). It is an essential gene in yeast; its deletion results in either lethality or severely impaired, temperature-sensitive growth26. A temperature-sensitive GWT1 mutant yeast strain (TSA655), which is lethal at 35o C is available (Euroscarf).
The mammalian and yeast orthologs share 36% amino acid identity (Uniprot). Both PIG-W and Gwt1 catalyze acylation of the inositol ring of GlcN-PI during GPI anchor biosynthesis. Gwt1 has been shown to complement PIG-deficient mutant cells10. GWT1 catalyzes a palmitoyl-CoA-dependent inositol acyl transfer, though other acyl chains of varying lengths may also be transferred. GWT1 mutant cells produce fewer GPI-anchored proteins, indicating that acylation is critical for their proper expression26. In both yeast and mammalian cells, inositol ring acylation enhances the efficiency of the first mannose residue attachment during GPI anchor biosynthesis but does not entirely block mannosylation in its absence10.
Worm (C. elegans)
Pigw-1 is the ortholog of the mammalian PIGW gene (WormBase). Pigw-1 is predicted to enable glucosaminyl-phosphatidylinositol O-acyltransferase activity and be involved in the GPI anchor biosynthesis and protein localization to plasma membrane.
Fly (D. melanogaster)
Pig-wb and pig-wa are both paralogs of the mammalian PIGW gene (FlyBase). These genes are predicted to function as glucosaminyl-phosphatidylinositol O-acyltransferases involved in the GPI anchor biosynthesis. They are also predicted to localize to the membrane and are expressed in the embryonic and larval midgut.
Human Cell Lines
PIGW -/- HEK293T knockout cells
PIGW-knockout cells (PIGW-/-) were generated from HEK293 cells using the CRISPR/Cas System and displayed a reduced expression of the endogenous GPI-anchored proteins CD55, CD59 and prion27.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a natural history study on all CDG types, including PIGW-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Organizations
Behind the Rainbow
GPI-anchor CDG Community Facebook Group
Mabry Syndrome Family Support Facebook Group
Publications
PIGW-CDG Scientific Articles on PubMed
Additional Resources
Kinoshita Lab and Edmondson lab (GPI anchor pathway/disorders researchers)
OMIM
IEMbase
Orphanet
Genetic Testing Registry
MedlinePlus
ClinVar
NIH
GeneCards
UniProt
References
- Chiyonobu, T., Inoue, N., Morimoto, M., Kinoshita, T. & Murakami, Y. Glycosylphosphatidylinositol (GPI) anchor deficiency caused by mutations in PIGW is associated with West syndrome and hyperphosphatasia with mental retardation syndrome. J. Med. Genet. 51, 203–207 (2014).
- Hogrebe, M. et al. A novel mutation in PIGW causes glycosylphosphatidylinositol deficiency without hyperphosphatasia. Am. J. Med. Genet. A 170, 3319–3322 (2016).
- Foskett, G. K. et al. Use of flow cytometry for diagnosis of epilepsy associated with homozygous PIGW variants. Pediatr. Neurol. 85, 67–70 (2018).
- Fu, L. ’na, Liu, Y., Chen, Y., Yuan, Y. & Wei, W. Mutations in the PIGW gene associated with hyperphosphatasia and mental retardation syndrome: a case report. BMC Pediatr. 19, 68 (2019).
- Peron, A. et al. PIGW-related glycosylphosphatidylinositol deficiency: Description of a new patient and review of the literature. Am. J. Med. Genet. A 182, 1477–1482 (2020).
- Fang, Z., Hu, C., Zhou, S. & Yu, L. PIGW-related glycosylphosphatidylinositol deficiency: A case report and literature review. Neurol. Sci. 45, 2253–2260 (2024).
- Meier, N. et al. Exome sequencing of fetal anomaly syndromes: novel phenotype-genotype discoveries. Eur. J. Hum. Genet. 27, 730–737 (2019).
- Feresin, A. et al. In-depth phenotyping of PIGW-related disease and its role in 17q12 genomic disorder. Biomolecules 14, 1626 (2024).
- Ronzoni, L. et al. Prenatal ultrasound findings associated with PIGW variants: One more piece in the FRYNS syndrome puzzle? PIGW-related prenatal findings. Prenat. Diagn. 42, 1493–1502 (2022).
- Murakami, Y. et al. PIG-W is critical for inositol acylation but not for flipping of glycosylphosphatidylinositol-anchor. Mol. Biol. Cell 14, 4285–4295 (2003).
- Kinoshita, T. & Fujita, M. Biosynthesis of GPI-anchored proteins: special emphasis on GPI lipid remodeling. J. Lipid Res. 57, 6–24 (2016).
- Kinoshita, T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biol. 10, 190290 (2020).
- Liu, Y.-S. & Fujita, M. Mammalian GPI-anchor modifications and the enzymes involved. Biochem. Soc. Trans. 48, 1129–1138 (2020).
- Englund, P. T. The structure and biosynthesis of glycosyl phosphatidylinositol protein anchors. Annu. Rev. Biochem. 62, 121–138 (1993).
- Watanabe, R. et al. The first step of glycosylphosphatidylinositol biosynthesis is mediated by a complex of PIG-A, PIG-H, PIG-C and GPI1. EMBO J. 17, 877–885 (1998).
- Orlean, P. & Menon, A. K. Thematic review series: lipid posttranslational modifications. GPI anchoring of protein in yeast and mammalian cells, or: how we learned to stop worrying and love glycophospholipids. J. Lipid Res. 48, 993–1011 (2007).
- Hong, Y. et al. Requirement of PIG-F and PIG-O for transferring phosphoethanolamine to the third mannose in glycosylphosphatidylinositol. J. Biol. Chem. 275, 20911–20919 (2000).
- Doerrler, W. T., Ye, J., Falck, J. R. & Lehrman, M. A. Acylation of glucosaminyl phosphatidylinositol revisited. Palmitoyl-CoA dependent palmitoylation of the inositol residue of a synthetic dioctanoyl glucosaminyl phosphatidylinositol by hamster membranes permits efficient mannosylation of the glucosamine residue. J. Biol. Chem. 271, 27031–27038 (1996).
- Maeda, Y. & Kinoshita, T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Prog. Lipid Res. 50, 411–424 (2011).
- Fujita, M. & Kinoshita, T. Structural remodeling of GPI anchors during biosynthesis and after attachment to proteins. FEBS Lett. 584, 1670–1677 (2010).
- Wu, T. et al. The Glycosylphosphatidylinositol biosynthesis pathway in human diseases. Orphanet J. Rare Dis. 15, 129 (2020).
- Murakami, Y. et al. Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome. J. Biol. Chem. 287, 6318–6325 (2012).
- Thompson, M. D. & Knaus, A. Rare genetic developmental disabilities: Mabry syndrome (MIM 239300) index cases and glycophosphatidylinositol (GPI) disorders. Genes (Basel) 15, 619 (2024).
- Dulac, O. What is West syndrome? Brain Dev. 23, 447–452 (2001).
- Knaus, A. et al. Characterization of glycosylphosphatidylinositol biosynthesis defects by clinical features, flow cytometry, and automated image analysis. Genome Med. 10, 3 (2018).
- Umemura, M. et al. GWT1 gene is required for inositol acylation of glycosylphosphatidylinositol anchors in yeast. J. Biol. Chem. 278, 23639–23647 (2003).
- Liu, Y. S. & Fujita, M. Mammalian GPI-anchor modifications and the enzymes involved. Biochemical Society Transactions. 48 (3), 1129-1138. (2020).
Show More