
Lay Summary
PIGS-CDG is a rare inherited condition that affects several parts of the body. To date, 13 cases of PIGS-CDG have been reported in the medical literature. PIGS-CDG is classified as a disorder of GPI-anchor biosynthesis. It is caused when an individual has mutations in both copies of their PIGS gene, which provides instructions for making a protein that participates in attaching proteins to GPI anchors. GPI anchors are molecules that “anchor” certain proteins to the cell surface. The PIGS protein is involved in stabilizing the machinery that attaches GPI anchors to proteins. Mutations in the PIGS gene cause defects in GPI-anchored proteins, making them unstable or unable to attach to the cell surface. PIGS-CDG is characterized by neurological and ophthalmological symptoms, including developmental delay, intellectual disability, seizures, low muscle tone, problems with balance and coordination and visual impairment. Some individuals diagnosed with PIGS-CDG may have a lower life expectancy due to complications. PIGS-CDG is usually diagnosed through genetic testing, however, testing for the absence of GPI-anchored proteins on certain blood cells can assist with screening for disorders of GPI anchor biosynthesis. Supplementation with vitamin B6 may reduce seizures in affected individuals, however, there are currently no approved treatments for PIGS-CDG. The present treatment is focused on the management of specific symptoms and preventing complications.
Overview
Phosphatidylinositol glycan class S congenital disorder of glycosylation (PIGS-CDG) is an autosomal recessive genetic disorder. The first reported case of PIGS-CDG was in 2013, and there have been 13 cases reported to date1–3. The PIGS gene encodes a subunit of the enzyme complex that catalyzes the attachment of GPI anchors to proteins during GPI-anchored protein biosynthesis. GPI anchors are an important mechanism of attachment for proteins to the cell surface, and these proteins are known as GPI-anchored proteins. Mutations in PIGS result in defects in GPI-anchored proteins, which are critical to the development and function of many organs, especially the nervous system4.
Symptoms of PIGS-CDG begin in infancy and the characteristic presentation includes delay, intellectual disability, developmental seizures, low muscle tone, ataxia, visual impairment and abnormalities1–3. PIGS-CDG may result in premature death; approximately half of the patients reported to date have not survived beyond early childhood. Although a definitive diagnosis can only be achieved through genetic sequencing, analyzing blood cells for an absence of GPI-anchored proteins can help diagnose disorders of GPI anchor biosynthesis While vitamin B6 supplementation has successfully reduced seizures in some patients, there are currently no therapies available for PIGS-CDG. Treatment is focused on managing symptoms and preventing complications.
Synonyms
- Glycosylphosphatidylinositol class S deficiency
- Developmental and epileptic encephalopathy 95; DEE95
- Glycosylphosphatidylinositol biosynthesis defect 18; GPIBD18
Inheritance
PIGS-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The PIGS gene encodes a subunit of the GPI transamidase (GPI-TA) complex, which is comprised of 5 protein subunits. This transamidase complex catalyzes the attachment of a protein to a GPI anchor in the endoplasmic reticulum (ER)5,6. Although PIG-S is an essential subunit for the enzymatic activity of the GPI-TA complex, its function is unclear5,7.
GPI-Anchor Protein Biosynthesis
GPI-anchored protein biosynthesis is one of the major glycosylation pathways that attach glycans to lipid molecules within cells. Many proteins, called GPI-anchored proteins, are attached to the cell surface by GPI anchors.
The GPI core structure consists of phosphatidylinositol (PI), glucosamine (GlcN), three mannose sugars (Man3), and three phosphoethanolamine (EtN-P) connected to each other in that sequence (Figure 1). GPI-anchored proteins are attached to the GPI by forming a bond between the EtN-P group of the GPI core and the C-terminus of newly synthesized proteins in the ER lumen5,6.
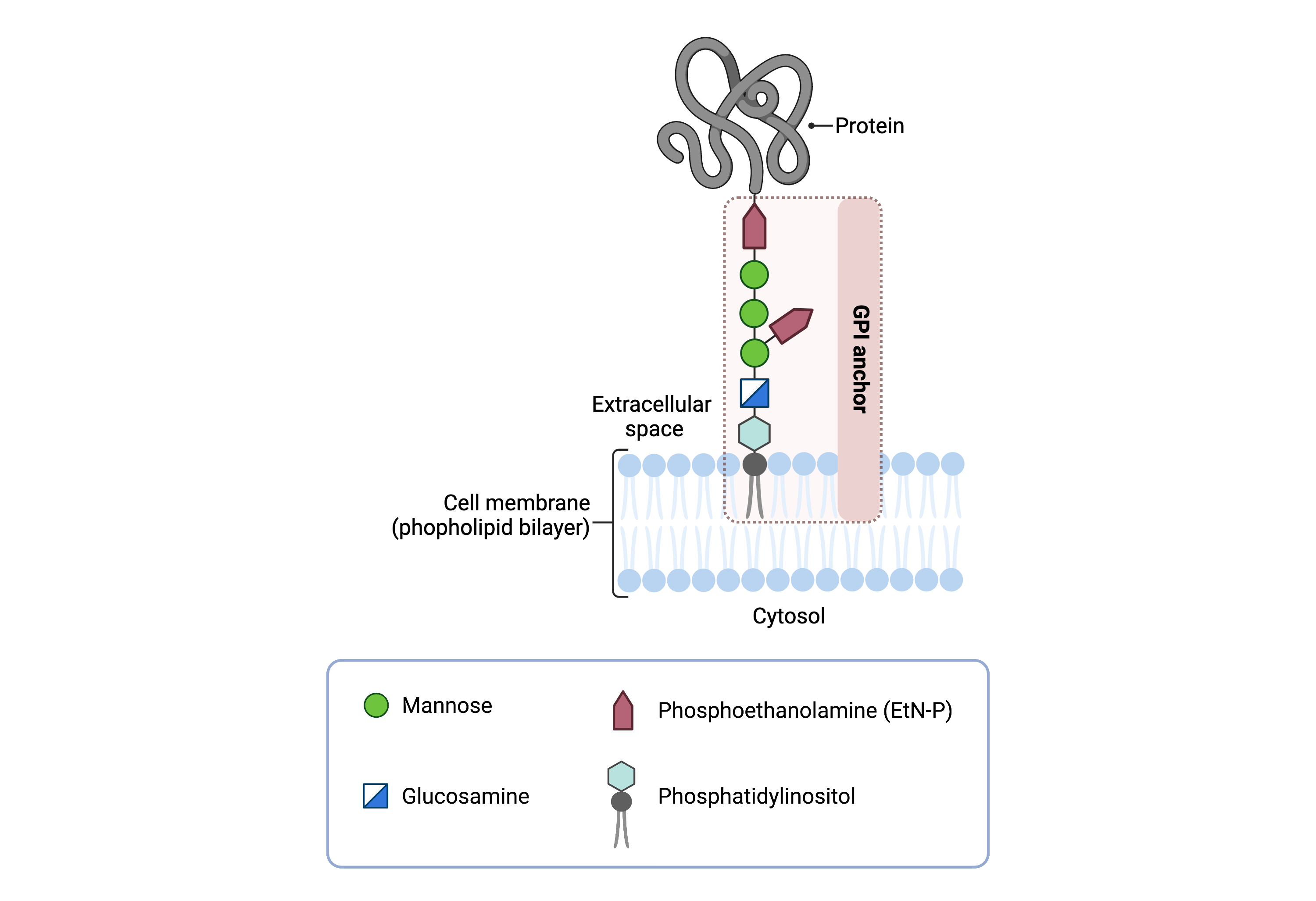
Figure 1.Overview of GPI-anchored protein structure.
The GPI anchor is added to proteins, generating GPI-anchored proteins. The GPI anchor section lodges into the membrane, attaching the protein to the membrane. The GPI anchor involves phosphatidylinositol, glucosamine, mannose and phosphoethanolamine.
The generation of GPI-anchored proteins is a multi-step process involving more than 30 enzymes and can be divided into the following steps (Figure 2):
- GPI anchor synthesis
- Protein attachment
- Lipid/glycan remodelling and transport
The first 2 steps occur in the ER, while step 3 occurs both in the ER and the Golgi. GPI anchor synthesis and protein attachment is largely carried out by a series of enzymes encoded by the PIG genes, while enzymes encoded by the PGAP genes facilitate remodelling of the GPI anchored protein.
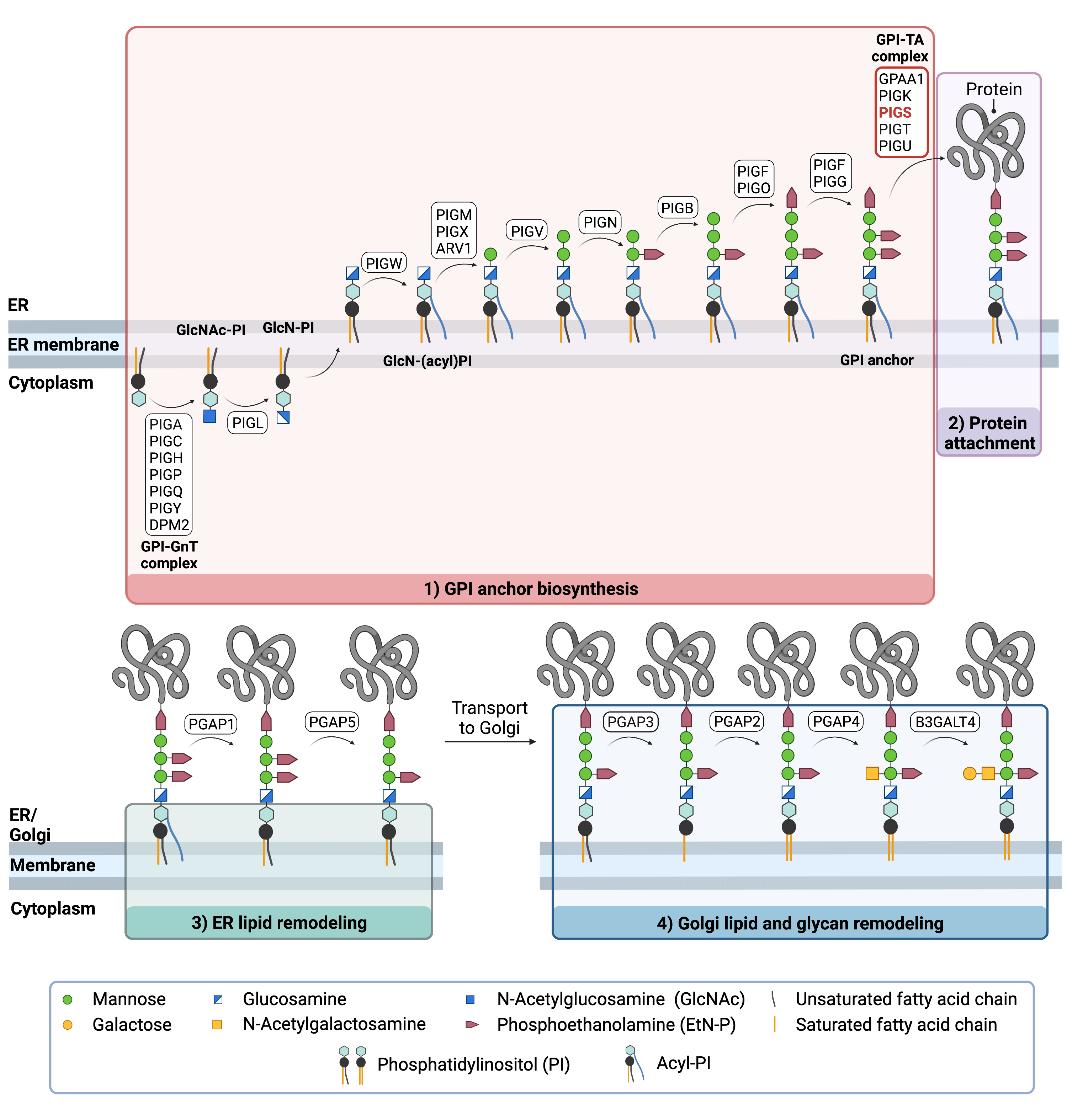
Figure 1. Overview of GPI-anchored protein biosynthesis and role of PIG-S.
GPI-anchored protein biosynthesis involves a series of enzymatic reactions. First, the GPI anchor core is built on the ER membrane, where enzymes modify the lipid and glycan portions, generating PI-GlcN-Man3-EtN-P. PIGS-S is one of 5 subunits of the GPI transamidase (GPI-TA) complex which catalyzes the attachment of the synthesized GPI anchor to proteins in the ER. Following GPI attachment, modifications are made to the lipid and glycan portions of the GPI-anchored protein in the ER and the Golgi.
GPI Anchor Synthesis
The first stage of GPI anchor biosynthesis involves the stepwise construction of the GPI anchor. N-acetylglucosamine (GlcNAc) is added to the lipid phosphatidylinositol (PI), generating GlcNAc-PI. GlcNAc-PI then has its acetyl (Ac) group removed from the GlcNAc sugar, generating glucosamine phosphatidyl inositol (GlcN-PI) which is transported into the luminal side of the ER5,6.
Within the ER, a fatty acid chain is attached to GlcN-PI. The final steps of the GPI anchor synthesis involve the sequential addition of 3-4 mannose sugars and three EtN-P groups by several PIG enzymes. The mannose residues are transferred from the ER-resident sugar donor, dolichol-phosphate mannose. The completed GPI anchor core, PI-GlcN-Man3-EtN-P, is then ready to be attached to a protein5,6.
Protein Attachment
The newly synthesized GPI anchor core is then transferred to a protein by GPI transamidase (GPI-TA). GPI transamidase is a large enzyme complex comprised of 5 proteins: where PIG-K and GAA1 are the catalytic subunits and PIG-U and PIG-T stabilize these proteins. PIG-S is essential for the catalytic activity of the GPI-TA complex; however, its exact function is not clear 7–9 Upon recognition of the signal sequence, GPI transaminase catalyzes the simultaneous cleavage of the signal sequence and attachment of the GPI anchor to the newly synthesized protein5,6.
Lipid/Glycan Remodelling and Protein Transport
After the protein has been attached to the GPI anchor, both the glycan and lipid portion of the anchor undergo modifications (referred to as remodelling) in the ER and Golgi by post-GPI attachment to protein (PGAP) enzymes or the glycosyltransferase B3GALT4. The complete GPI-anchored protein is then transported to the plasma membrane where it associates with other GPI-anchored proteins in lipid rafts 5,10–13.
Disease Mechanism
As PIG-S is essential for the catalytic activity of the GPI transamidase complex, mutations in the PIGS gene render the complex unable to transfer the GPI anchor to a precursor protein bearing a GPI-attachment signal sequence 1,14. As a result, proteins which require a GPI anchor may not be attached to the cell surface and perform their function. GPI-anchored proteins have several critical functions in the cell such as adhesion molecules, receptors, and enzymes in signal transduction pathways, and are also important in embryogenesis and neurogenesis.
Mutations
The PIGS gene is located on chromosome 17 (17q11.2). To date, 12 variants in the PIGS gene have been reported in the literature, including 4 missense, 3 nonsense, 2 splice-site variants, 1 insertion, 1 deletion, and 1 duplication. In the 4 deaths that have been reported, 3 of the patients had a unique missense mutation each, and were either homozygous for the variant (c. 1070 G>A, p. Gly357Asp; c.986 C>G, p. Pro329Arg), or were compound heterozygous with the missense mutation (c.923A>G, p.Glu308Gly) accompanied by a splice-site variant (c.468 + 1G-C)1,3. One reported death was of a child aged 15 months, who was compound heterozygous for a nonsense mutation and a duplication2.
Signs & Symptoms
Clinical Presentation
Individuals with PIGS-CDG typically develop signs and symptoms during infancy. PIGS-CDG is primarily characterized by severe global developmental delay, epilepsy, low muscle tone and visual impairment. Symptoms of PIGS-CDG include1–3:
- Neurological– global developmental delay, low muscle tone (hypotonia), epileptic seizures, brain abnormalities, absent speech
- Ophthalmological – involuntary eye movements (nystagmus), blindness originating from brain defects
- Dysmorphic features – small head size (microcephaly), facial dysmorphism (arched eyebrows, thickened helices, broad tongue), joint abnormalities, short fingers and toes (brachydactyly)
Less common symptoms of PIGS-CDG include hearing loss and kidney malformations.
Biomedical Abnormalities
Mildly elevated alkaline phosphatase levels have been reported in two patients, however, levels appear to be normal in the majority of patients reported to date1–3.
Classification
PIGS-CDG is classified as a disorder of GPI-anchor biosynthesis.
Diagnosis
GPI-related CDG should be considered in individuals presenting with early onset severe seizure disorders and dysmorphic facial features, even if transferrin and total N-glycan analysis are normal. As currently available screening tests for CDG will not reliably detect PIGS-CDG, diagnosis is typically achieved through genetic testing, either as part of an epilepsy panel or whole exome sequencing. PIGS-CDG may also be primarily screened for by analyzing surface GPI-anchor proteins on blood cells by flow cytometry.
GPI Anchored Protein Flow Cytometry
Individuals with PIGS-CDG lack GPI-anchor proteins on the surface of their granulocytes, a type of white blood cell.
Biomarkers
No biomarkers for PIGS-CDG have been reported.
Prognosis
The prognosis of PIGS-CDG may vary depending on the severity of an individual’s symptoms. The broad clinical spectrum may result in premature death in some patients as a result of respiratory complications. As of 2020, the oldest patient reported in the literature is 8 years old1–3.
Management
Management of symptoms may include combinations of physical therapy, occupational therapy, and palliative measures. The function of GPI-anchored proteins are implicated in vitamin B6 and folate transport, nucleotide metabolism, and lipid homeostasis. In one study, two patients were treated with pyroxidine and folinic acids which provided some benefit in seizure control3 . A ketogenic diet has previously been reported to improve outcomes in other cases of PIGA-CDG15 and may be considered for individuals with PIGS-CDG3.
Therapies
There are currently no therapies available for PIGS-CDG.
Research Models
Several research models are available to study PIGS-CDG including yeast and a human cell line.
Yeast (S. cerevisiae)
GPI17 is the yeast orthologue of PIGS. GPI17 deletion yeast display a reduced transfer of GPI anchors to proteins and an accumulation of GPI precursors (SGD)8.
Mouse (M. musculus)
Pigs KO mouse embryonic carcinoma F9 cells
The Pigs gene in the mouse embryonic carcinoma cells was knocked out by homologous recombination. Pigs KO F9 cells were impaired in their ability to transfer GPI to proteins8.
Human Cell Lines
HEK293 PIGS-/- knockout cells
A HEK293 knockout library of genes involved in GPI biosynthesis in HEK293 cells was generated for functional studies. Knockout of PIGS in HEK293 cells results in the inactivation of the GPI-TA complex and GPI-anchored proteins are not synthesized (as measured by surface CD55 and CD59 expression)16.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including PIGS-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Publications
PIGS-CDG Scientific Articles on PubMed
Additional Resources
IEMbase
OMIM
Orphanet
Genetic Testing Registry
ClinVar
NIH
GeneCards
UniProt
Marrvel
References
- Nguyen, T. T. M. et al. Mutations in PIGS, Encoding a GPI Transamidase, Cause a Neurological Syndrome Ranging from Fetal Akinesia to Epileptic Encephalopathy. Am. J. Hum. Genet. 103, 602–611 (2018).
- Zhang, L. et al. Compound Heterozygous PIGS Variants Associated With Infantile Spasm, Global Developmental Delay, Hearing Loss, Visual Impairment, and Hypotonia. Front. Genet. 11, (2020).
- Efthymiou, S. et al. Expanding the phenotype of PIGS-associated early onset epileptic developmental encephalopathy. Epilepsia 62, e35–e41 (2021).
- Paulick, M. G. & Bertozzi, C. R. The Glycosylphosphatidylinositol Anchor: A Complex Membrane-Anchoring Structure for Proteins. Biochemistry 47, 6991 (2008).
- Kinoshita, T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biology vol. 10 190290 (2020).
- Liu, Y. S. & Fujita, M. Mammalian GPI-anchor modifications and the enzymes involved. Biochemical Society Transactions vol. 48 1129–1138 (2020).
- Liu, S. S. et al. Functional Analysis of the GPI Transamidase Complex by Screening for Amino Acid Mutations in Each Subunit. Mol. 2021, Vol. 26, Page 5462 26, 5462 (2021).
- Ohishi, K. PIG-S and PIG-T, essential for GPI anchor attachment to proteins, form a complex with GAA1 and GPI8. EMBO J. 20, 4088–4098 (2001).
- Kawaguchi, K., Sato, T., Kondo, S., Yamamoto-Hino & Goto, S. Stability of the transamidase complex catalyzing GPI anchoring of proteins. Biochem. Biophys. Res. Commun. 512, 584–590 (2019).
- Liu, Y. S. & Fujita, M. Mammalian GPI-anchor modifications and the enzymes involved. Biochemical Society Transactions vol. 48 (2020).
- Maeda, Y. & Kinoshita, T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Prog. Lipid Res. 50, 411–424 (2011).
- Kinoshita, T. & Fujita, M. Biosynthesis of GPI-anchored proteins: Special emphasis on GPI lipid remodeling. Journal of Lipid Research vol. 57 6–24 (2016).
- Maeda, Y. et al. Fatty Acid Remodeling of GPI-anchored Proteins Is Required for Their Raft Association D. Mol. Biol. Cell 18, 1497–1506 (2007).
- Ohishi, K., Inoue, N. & Kinoshita, T. PIG-S and PIG-T, essential for GPI anchor attachment to proteins, form a complex with GAA1 and GPI8. EMBO J. 20, 4088–4098 (2001).
- Joshi, C. et al. Ketogenic diet – A novel treatment for early epileptic encephalopathy due to PIGA deficiency. Brain Dev. 38, (2016).
- Liu, S. S. et al. A knockout cell library of GPI biosynthetic genes for functional studies of GPI-anchored proteins. Commun. Biol. 4, (2021).