
Lay Summary
NGLY1 congenital disorder of deglycosylation (NGLY1-CDDG), also commonly referred to as NGLY1 deficiency, is a rare inherited condition that affects many systems in the body. To date, over 100 patients with NGLY1-CDDG have been reported. It is classified as a congenital disorder of deglycosylation (CDDG). NGLY1-CDDG is caused when an individual has mutations in both copies of their NGLY1 gene, which provides instructions for making an enzyme that removes sugar chains (glycans) from proteins. The removal of sugar chains, known as deglycosylation, by the NGLY1 enzyme plays an essential role in the breakdown of down abnormal proteins. As well, some properly folded proteins require removal of their glycan chain by NGLY1 before they can function properly. Mutations in the NGLY1 gene prevent misfolded proteins from being properly broken down, causing them to build up in cells. Symptoms of NGLY1-CDDG begin at infancy and are characterized by neurodevelopmental delay, reduced or inability to secrete tears, liver abnormalities, reduced muscle tone and abnormal involuntary movements, and damage to nerves in the limbs and extremities. Typical screening tests used for congenital disorders of glycosylation cannot reliably diagnose NGLY1-CDDG and a definitive diagnosis is achieved through genetic testing. Potential biomarkers for NGLY1-CDDG have been identified which may assist in diagnosis. Pharmaceutical drugs, gene therapy and enzyme replacement therapies are in development to treat NGLY1-CDDG.
Overview
N-glycanase I congenital disorder of deglycosylation (NGLY1-CDDG) is a rare autosomal recessive genetic disorder that arises from defects in the NGLY1 (N-glycanase 1) gene and is one of only two congenital disorders of deglycosylation currently known1–3. The NGLY1 gene encodes an enzyme (NGLY1) that is responsible for cleaving (deglycosylating) the sugar chains (glycans) from N-glycosylated proteins (N-glycoproteins). Deglycosylation serves several functions. It is the first step in the degradation and recycling misfolded N-glycoproteins, and proper NGLY1 function ensures that misfolded N-glycoproteins do not accumulate in cells and cause toxicity. As well, deglycosylation by NGLY1 is necessary to active some N-glycoproteins that affect a wide range of processes including homeostasis and embryonic development. Mutations in NGLY1 result in NGLY1 being unable to properly cleave N-glycans, which has multiple down-stream effects on many cellular processes.
The first reported case of NGLY1-CDDG was in 2012 and to date more than 60 patients have been clinically described, with over 100 diagnoses made overall1,2,4–23. Symptoms of NGLY1-CDDG begin at infancy and present as a neurodevelopmental disorder with multisystem involvement including developmental delays, liver abnormalities, reduced ability to secrete tears, movement disorder and peripheral neuropathy. A definitive diagnosis can only be obtained through whole genome sequencing; however, screening tests for biomarkers unique to NGLY-CDDG may aid in diagnosis and screening. Although there is currently no available treatment for NGLY1-CDDG, gene, small molecule and dietary therapies are currently under development.
Synonyms
- NGLY1-related congenital disorder of deglycosylation
- NGYL1-related disorder
- NGLY1 deficiency
- Congenital disorder of glycosylation Iv
- CDG Iv
Inheritance
NGLY1-CDDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The NGLY1 gene encodes the enzyme N glycanase I (NGLY1), which is primarily localized in the cytoplasm and is responsible for de-glycosylation of various N-glycoproteins24. De-glycosylation by NGLY1 involves cleaving the bond between the glycan chain and the asparagine it was attached to on the N-glycoprotein, which generates a free N-glycan and concomitantly converts the asparagine (Asn; D) residue into an aspartic acid (Asp; N) residue (Figure 1). NGLY1 also plays a key role in the degradation of misfolded N-glycoproteins through ER-associated degradation, which require removal of their glycan chain before they are degraded in the proteasome. The post-transcriptional amino acid editing (Asn to Asp) affects the charge and structure of the protein, and some glycoproteins require this modification to gain full functionality3. For example, the transcription factor Nuclear Factor Erythroid 2 Like 1 (NRF1/NFE2L1) only gains full functionality once its glycan chain is removed by NGLY13,25–27. Due to the many proteins that NGLY1 interacts with and their downstream effects, normal NGLY1 function is important for many other processes including proper mitochondrial function, regulation of water channels, proteasome and cellular energy homeostasis, ion transport, inflammation, lipid metabolism and programmed cell death.
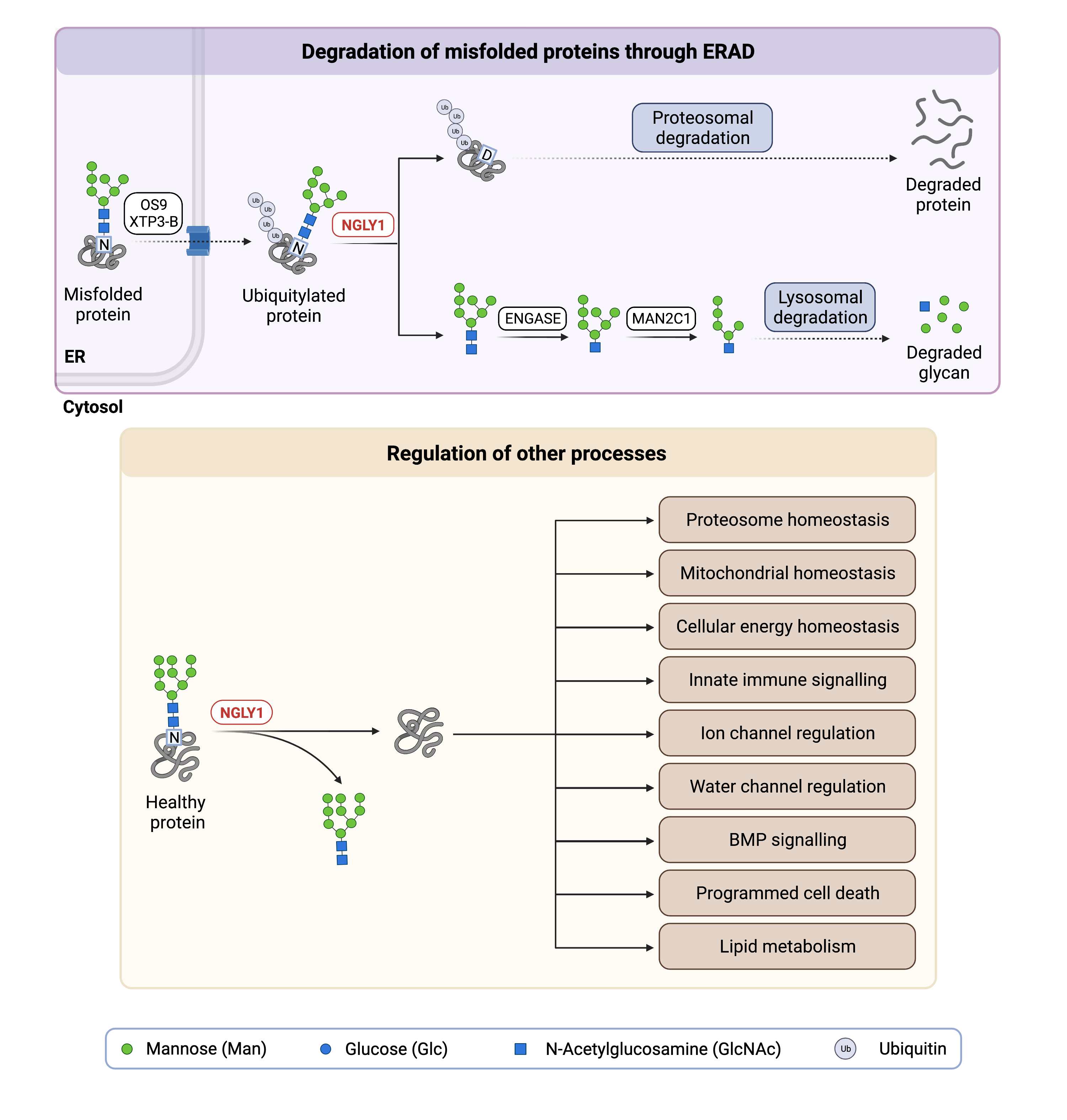
Figure 1. Role of NGLY1 in glycosylation and regulation of other processes.
N glycanase I (NGLY1) is a deglycosylation enzyme, where NGLY1 removes N-glycans from proteins. NGLY1 deglycosylation of N-glycoproteins is involved in protein degradation (ERAD) and the regulation of various processes.
ER-Associated Degradation
The removal of N-glycan chains from misfolded N-glycoproteins by NGLY1 is a key step in ER-associated degradation (ERAD). ERAD is a quality control mechanism for newly synthesized proteins in the endoplasmic reticulum (ER) that ultimately results in misfolded proteins being transported to the proteasome where they are broken down (Figure 1). Misfolded glycoproteins are first detected by lectins in the ER and translocated out of the ER into the cytosol. Before N-glycoproteins are degraded by the proteasome they are deglycosylated by NGLY1. NGLY1 cleaves the glycan from the asparagine residue to which it is attached, generating a free glycan and simultaneously converting the asparagine residue into an aspartate residue1,28. The free glycan is further broken down by another enzyme, endo-β-N-acetylglucosaminidase (ENGase). However, the role of NGLY1 in ERAD continues to be investigated as a loss of NGLY1 does not result in widespread ERAD defects and de-glycosylation of substrates is still observed24.
Activation and Regulation of NRF1
Many of the processes regulated by NGLY1 are mediated by NRF1, a transcription factor that is activated by the removal of its glycan chain by NGLY1. NRF1 regulates the expression of various genes and plays complex roles in mitochondrial, proteasomal and lipid homeostasis, inflammation, and a type of programmed cell death called ferroptosis3,25–27,29.
Regulation of Other Process
NGLY1 activity affects many other proteins, pathways, and processes in the body. For example, it activates tissue-specific bone morphogenetic protein (BMP), a signalling protein that plays an important role in embryonic development30,31. As well, the sodium-potassium-chloride cotransporters (NKCC1/2) are involved in fluid secretion and depend on normal NGLY1 activity32. NGLY1 also plays an important role in energy homeostasis by maintaining adequate levels of AMP-activated protein kinase α (AMPKα), a protein that promotes energy production when cells are low on energy33.
Finally, NGLY1 also regulates multiple aquaporin channels at the transcriptional level via the Aft1/Creb1 signaling pathway through a non-enzymatic mechanism that remains incompletely understood34. Aquaporins are membrane proteins that are channels for water and small solutes to cross the cell membrane and are necessary for the formation of tears. Overall, proper NGLY1 function impacts multiple systems by activating or regulating proteins involved in homeostasis and development.
Disease Mechanism
NGLY1 affects the breakdown and/or function of N-glycoproteins involved various biological processes, and loss of NGLY1 function directly and indirectly impacts many key processes in the body. NGLY deficiency directly impacts ERAD, leads to reduced NRF1 function, causes accumulation of toxic protein aggregates, increased proteasomal and mitochondrial stress, dysregulation of lipid metabolism, and reduced aquaporin expression; some of these effects are likely due to inadequate activation of transcription factor NRF1.
Dysregulation of ERAD
ERAD is an essential process by which misfolded proteins can be broken down and recycled. Dysregulation of ERAD can lead to the accumulation of toxic protein aggregates that impair proteasome function, which further worsens ERAD dysregulation.
An important step of ERAD is the cleavage of the glycans from misfolded glycoproteins by NGLY1. These glycans are then further deglycosylated by ENGase before ultimately being broken down in the lysosome (Figure 1). However, if misfolded N-glycoproteins accumulate due to reduced NGLY1 activity, ENGase will remove the glycan chain but leave the innermost GlcNAc residue of the glycan attached to the protein. These N-GlcNAc proteins are prone to forming toxic aggregates in the cell and impede normal ERAD28. Furthermore, in the absence of NGLY1, proteins that would normally be processed by ERAD can be abnormally ubiquitinated by ubiquitin ligase; these abnormally ubiquitinated proteins impair the proteasome25. Overall, proteasomal impairment and ERAD dysregulation lead to toxic protein aggregates and contribute to the phenotype observed in NGLY1 deficiency.
Dysregulation of NRF1
NRF1 regulates the expression of various genes and is particularly important for proteasomal and mitochondrial homeostasis and resistance to a form of cell death called ferroptosis29. During proteasomal stress, NRF1 will upregulate the expression of proteosome subunits to increase the processing capacity of the proteosome25,26. If NRF1 is not activated by NGLY1 and is inappropriately ubiquitinated, this response is impeded and can lead to accumulation of misfolded proteins and cellular toxicity25.
As well, NRF1 is important for recycling of damaged mitochondria (mitophagy), and deficiency in NGLY1 reduces the ability of mitochondria to produce energy (oxidative phosphorylation) and increases oxidative stress in the mitochondria27,35. Poor mitochondrial function in the liver likely plays a role in the liver phenotype displayed by NGLY1-deficient patients6. Damaged mitochondria can leak mitochondrial DNA and RNA into the cytosol, leading to persistent activation of an innate immune response pathway which has been associated with autoimmune and autoinflammatory diseases, but may also provide increased resistance to viral infection27,36. The lack of activation of NRF1 in NGLY1-deficient patients likely explains some symptoms of NGLY1-CDDG.
Dysregulation of Other Processes
NGLY1 activity impacts several other important pathways in the body, including the Bone Morphogenic Protein (BMP) signalling cascade, lipid metabolism in the liver, and aquaporin expression. BMP signalling regulates many aspects of embryonic development in humans, and deficiency in NGLY1 causes alterations in BMP signalling that may contribute to some phenotypes of NGLY1-CDDG such as retinal abnormalities, small feet and hands, delayed bone age and osteopenia, and chronic constipation30,31.
As well, NGLY1 has been shown to play a role in lipid metabolism in the liver. Hepatocyte-specific Ngly1-deficient mice presented with elevated liver transaminases, fatty liver, and increased lipid droplet accumulation under a high fructose diet, which suggests that lipid metabolism in the liver is dysregulated when Ngly1 is deficient/absent37.
Aquaporins are membrane proteins that are channels for water and small solutes to cross cell membranes. NGLY1 regulates the expression of multiple aquaporin channels, and in NGLY1-deficiency the mRNA and protein levels of various aquaporins are reduced34. Furthermore, NKCC1 is an ion transporter involved in fluid secretion that has reduced activity in the absence of NGLY132. Reduced aquaporin expression and NKCC1 activity may contribute to the reduced ability or inability to secrete tears and gastrointestinal symptoms (chronic constipation) in NGLY1-CDDG patients16.
Mutations
The NGLY1 gene is found on chromosome 3 (3p24.2), and at least 50 different pathogenic variants have been identified including nonsense, missense, frame shifts, and splicing mutations4,9,10,16,22,23. Most pathogenic variants are nonsense and small deletion mutations that result in loss-of-function. The nonsense c.1201A>T(p.R401X) mutation is the most common mutation, accounting for 20% of observed pathogenic alleles in patients, and is associated with more severe disease4,10.
Signs & Symptoms
Clinical Presentation
Individuals with NGLY1-CDDG typically develop signs and symptoms during infancy. NLGY1-CDDG is characterized by neurodevelopmental disorder with multisystem involvement in which individuals most commonly exhibit global development mental delay and/or intellectual disability, abnormal involuntary movement, absent or reduced ability to produce tears, seizures, liver problems and polyneuropathy. The characteristic clinical presentations of NGLY1-CDDG include3,9,13,16,19,21,38:
- Neurological – developmental delay, intellectual disability (ranging from slightly below average IQ to completely non-verbal), decreased muscle tone (hypotonia), reduced sensation and weakness in arms and legs (peripheral neuropathy), absent sweat response, abnormal involuntary movements (hyperkinetic movement disorder), smaller head size (microcephaly), brain imaging and activity abnormalities, and seizures ranging in severity from sub-clinical to severe
- Musculoskeletal – abnormal curvature of the spine (scoliosis) that may worsen over time, small hands and feet, tightening of muscles, tendons, ligaments or skin (contractures)21
- Growth/feeding problems – failure to thrive which includes poor growth in the womb, difficulty putting on weight due to feeding problems and difficulty meeting motor skills milestones in the first 18 months. Feeding difficulties arise due to difficulty chewing and swallowing solid foods, and difficulty holding food in hands
- Breathing problems: central and/or obstructive sleep apnea.
- Liver involvement – accumulation of fat in the liver (steatosis) and liver scarring (fibrosis and cirrhosis)6,7,11,12,18
- Ophthalmological – reduced or absent ability to secrete tears (alacrima/hypolacrima), crossed eyes (strabismus), sores or scarring on the cornea, loss of control of voluntary eye movement
Biochemical Abnormalities
Biochemical abnormalities may include elevated liver proteins (transaminases and alpha-fetoprotein) in the blood that typically resolves in older patients, general aminoaciduria (especially in older patients), and mild to moderately elevated blood lactate9,19,38.
Classification
NGLY1-CDDG is a disorder of deglycosylation.
Diagnosis
NGLY1 deficiency should be considered in any multisystemic disorder, especially when a patient presents with global developmental delay, a hyperkinetic movement disorder, and/or alacrima4,5,9,19,21. Although diagnosis of NGLY1-CDDG may be suspected based on presentation of symptoms and a detailed patient history, genetic testing is the only definitive diagnostic test. The typical serum screening tests used for CDG to detect N- or O-glycan abnormalities, (i.e., transferrin analysis or apolipoprotein C-III analysis, or total glycan profiling), will not reliably detect NGLY1-CDDG. There are potential blood and urine biomarkers for NGLY1-CDDG that may aid in diagnosis.
Biomarkers
Several potential diagnostic and therapeutic biomarkers have been discovered for NGLY1-CDDG.
Aspartylglycosamine
Aspartylglycosamine is a potential biomarker tested through dried blood spot, and patients with NGLY1-CDDG have increased levels of aspartylglycosamine. In the absence of adequate NGLY1 activity, ENGase will cleave act on misfolded glycoproteins and leave a single N-acetylglucosamine (GlcNAc) sugar attached to a protein asparagine residue. When the protein is further degraded, this results in the formation of aspartylglycosamine39.
GlcNAc-Asn (GNA)
GlcNAc-Asn (GNA) is a potential biomarker found in urine, plasma, and dried blood spots of NLGY-CDDG patients40. When ENGase acts on misfolded N-glycoproteins in the absence of NGLY1, it generates proteins with a single acetylglucosamine (GlcNAc) sugar attached to the asparagine (Asn) residue. When the protein is further degraded and recycled into amino acids, some of this asparagine may retain the acetylglucosamine sugar, thus forming GlcNAc-Asn.
Asparagine-N (Asn-N)
Neu5Ac1Hex1GlcNAc1-Asn, also called asparagine-N (Asn-N) is a potential oligosaccharide biomarker for NGLY1-CDDG that has been shown to be elevated in urine samples from patients with NGLY1-CDDG using MALDI-TOF mass spectrometry41. This oligosaccharide fragment likely results from additional modification of GlcNAc-Asn, which is formed by the inappropriate action of ENGase on glycoproteins in the absence of NGLY1.
Prognosis
Prognosis of NGLY1-CDDG may vary depending on the severity of an individual’s symptoms. Most patients survive into early adulthood, however death during infancy of NGLY1-CDDG patients has been reported with an unclear cause. Early death in childhood or adolescents has also been caused by seizures, respiratory failure, and adrenal insufficiency20,38.
Management
Management of NGLY-CDDG requires a multidisciplinary team and may include feeding therapy, supportive therapy for developmental and cognitive problems, and standard treatments for other manifestations such as hearing loss, scoliosis, sleep apnea, and constipation38. Seizures may be managed in some patients through medication10,15,19. Lubricating eye drops and ointments should be used to treat alacrima. Patients should have access to adequate water and a cool environment to avoid overheating due to hypohydrosis; a cooling vest may help patients who live in hot climates38.
Therapies
There are currently no approved treatments available for NGLY1-CDDG, but dietary supplements, small molecule drugs, gene therapy, and enzyme replacement therapy are currently being explored in pre-clinical and clinical studies.
Gene Therapy
Gene therapy aims to treat the disorder at the gene level by modifying or replacing the disease-causing gene within a patient. GS-100, a gene therapy developed by Grace Science LLC, demonstrated efficacy in improving behavioural phenotypes and disease biomarkers in a rat model of NGLY1 deficiency61. Grace Science LLC is now conducting a clinical study to assess the safety and efficacy of intracerebroventricular (ICV) administration of a GS-100 in patients with NGLY1 deficiency. The study is currently recruiting participants. For more information on the study and how to enrol, visit the study website here
GlcNAc Dietary Supplementation
A study by Owings et al. 2018 demonstrated that GlcNAC synthesis is dysregulated in a fly model of NGLY1 deficiency. In the absence of the NGLY1 enzyme, misfolded proteins with GlcNAc still attached, accumulate in the cytoplasm and deplete the cell's supply of the GlcNAc sugar. In NGLY1 knockdown flies, GlcNAc supplementation partially rescued the lethality phenotype. A clinical study is being conducted to assess if dietary GlcNAc supplementation improves tear production in patients with NGLY1-CDDG. The study will soon be recruiting participants.For more information on the study and how to enrol, visit the study website here
Small Molecule Therapies
Grace Science LLC is conducting drug screens to identify small molecule treatments for NGLY1-CDDG. Travere Therapeutics id developing assays for small molecule high-throughput screening in an effort to better understand the biology of the disorder and identify potential compounds that can be developed as a therapeutic treatment.
Therapeutic Targets
Other potential therapeutic targets that have been identified include NRF2, ENGase, and FBS2. NRF2 is a transcriptional activator that can compensate for reduced NRF1 activity; compounds that induce NRF2 activity could thus mitigate the effects of decreased NRF1 function in NGLY1 deficiency42. The improper action of ENGase on misfolded proteins in the absence of NGLY1 is related to the development of toxic protein aggregates, and thus ENGase inhibitors may have therapeutic benefit for NGLY1 deficiency43. FBS2 is the subunit of the ubiquitin ligase complex (involved in the ERAD pathway) that recognizes the N-glycans of cytosolic glycoproteins. In the absence of NGLY1, some glycoproteins are abnormally ubiquitinated and impair the proteosome; inhibition of FBS2 rescues the lethality of NGLY1 deficiency in mice and may similarly benefit NGYLY1-CDDG patients25.
Research Models
Numerous NGLY1 research models have been generated including human cell lines and organoids, fly, worm, and rodent models3,16,44–46.
Worm (C. elegans)
The Png-1 gene in nematodes is orthologous to NGLY1 in humans. Png-1 knockout worms have reduced life span, neuronal branching defects, abnormal mitochondrial function, and are hypersensitive to proteasome inhibition35. The mechanism by which PNG-1 activates the transcription factor SKN1-A (C. elegans orthologue of NRF1) and its regulation of various pathways has been investigated in this model42,45,47.
Fly (D. melanogaster)
Drosophila gene Png1 (PNGase-like, CG7865, FBgn0033050) is orthologous to human NGLY1, and several models have been used to study NGLY1-CDDG (FlyBase)46.
RNAi Png1 knockdown models
RNAi knockdown of Png1 to mimic the loss of function of NGLY1 resulted in developmental delay and lethality at both the larval and adult stage. Dysregulation of GlcNAc synthesis, NRF1 dysfunction, and an adaptive upregulation of the heat shock response were all noted and suggested as targets for therapeutic intervention for NGLY1-CDDG48. This model was modified to explore phenotypic variability in NGLY1 deficiency by crossing the NGLY1 deficient model onto genetically diverse strains. This model identified the ion transporter Ncc69 (orthologous to human NKCC1/2) as modifying survival in NGLY1 deficiency; flies that were doubly knocked down for NGLY1 and Ncc69 demonstrated complete lethality while glial cell specific double knockdown demonstrated a unique seizure phenotype32. Other models show reduction in AMP-activated protein kinase (AMPK) leading to impaired energy metabolism, impaired gut peristalsis and emptying, and death33. An RNAi knockdown model was used to study the impact of NGLY1 deficiency on BMP4 signalling30.
Pngl PL (Png1 nonsense mutation) model
A fly model homozygous for a loss of function mutation in Png1 was created, showing global development delay, early lethality/decreased life span, small body size, and sterility; neuroendocrine abnormalities contribute to the global development delay phenotype, as Png1 is required for NRF1 function in the Drosophila neuroendocrine axis49. This model was modified to perform a repurposed drug screen, which identified therapeutic potential in activating NRF2, activating anti-inflammatory pathways, and boosting catecholamine levels in the brain42.
Mouse (M. musculus)
The Ngly1 gene in mice is orthologous to human NGLY1, and several mouse models have been generated to investigate NGLY1-CDDG44.
Mouse embryonic fibroblasts (MEFs)
Ngly1-deficient mouse embryonic fibroblasts (MEFs) display altered protein expression and function, impaired mitochondrial physiology, and reduced mitochondrial respiration30–33,35. Ablation of Ngly1 in MEFs also causes an accumulation of N-GlcNAc protein due to impaired ERAD processes, which can be rescued by additional knockout of the Engase gene28. ENGase can cleave the bond between the sialic acid residues present on the glycan chain of N-glycoproteins. In the absence of Ngly1, ENGase will act on misfolded N-glycoproteins and generate N-GlcNAc proteins that are prone to aggregation and can cause dysregulation of ERAD; this dysregulation is prevented in MEFs lacking both Ngly1 and ENGase. Ngly1-deficient MEFs also show reduced aquaporin1 mRNA and protein expression, reduced function of the ion transporter NKCC1, and resistance to hypotonic lysis, information that could be valuable in the study of defects in secretory epithelium function and alacrima noted in NGLY1-CDDG patients32,34.
Ngly1-/-constitutive knockout (C57BL/6)
Ngly1 knockout mice derived from the C57BL/6 mouse strain are embryonic lethal between E16.5 and birth and show ventricular septal defects50.
Ngly1-/- constitutive knockout (JF1)
Similar to mice from the C57BL/6 strain, Ngly1 whole body knockout is embryonic lethal in mice from the Japanese fancy mouse 1 (JF1) strain51.
Ngly1-/- constitutive knockout (JF1/B6F1)
Whereas Ngly1 knockout is lethal in both C57BL/6 and JF1 strains, an Ngly1 knockout mouse model prepared by crossing those strains (JF1/B6F1) is non-embryonic lethal51. The mice show developmental delay, motor dysfunction, accumulation of ubiquitinated proteins in the CNS, and increased levels of aspartylglycosamine in urine and plasma, similar to human patients with NGLY1-CDDG. This makes JF1/B6F1 Ngly1 knockout mice useful for preclinical testing of therapeutics and studying the mechanisms behind NGLY1 deficiency.
Ngly1/Engase double knockout (C57BL/6)
The embryonic lethality of C57BL/6 mice lacking the Ngly1 gene was partially rescued by the additional knockout of the Engase gene50. However, double knockout mutants present with a bent spine, hind-limb clasping, front-limb shaking, and reduced body weight.
Ngly1/Fbs2 double knockout
Fbs2 encodes a subunit of the ubiquitin ligase complex that recognizes glycoproteins. In the absence of Ngy1, the ubiquitin ligase complex causes abnormal ubiquitination of the N-glycoprotein NRF1 in inhibition of proteasomal activity. In mice lacking Ngly1, further knockout of the Fbs2 gene rescues the lethality phenotype and the mice survive to adulthood with normal motor function, indicating Fbs2 as a potential target for future therapeutics25.
Ngly1 hepatocyte-specific conditional knockout
Liver-specific knockout of Ngly1, generated using cre-loxP system, caused abnormal hepatocyte nuclear size and morphology with aging37. Stress to the liver, in the form of a high fructose diet, caused elevation of liver transaminases and lipid droplet accumulation. These mice could serve a model for the assessment of therapies for treating NGLY1-CDDG.
Ngly1em4Lutzy null mutant
Null mutant embryos demonstrated a significant decrease in BMP4 signalling in the heart and brain, ventricular septal defects, decrease in heart size and myocardial trabeculae, and significant abnormalities in parts of the brain (4th ventricle choroid plexus)30.
Rat (R. Rattus)
Ngly1-/- constitutive knockout
The Ngly1 gene in rats is orthologous to human NGLY1. Rats with complete Ngly1 deficiency show neurological symptoms, developmental delay, movement disorder, somatosensory impairment, and scoliosis, consistent with clinical presentation of human patients with NGLY-CDDG52,53. Accumulation of cytoplasmic ubiquitinated proteins in neurons in the thalamus and spinal cord were also observed. The similarity between clinical features in human patients and Ngly1 full body knockout rats make rat models useful for studying the mechanism of NGLY1-CDDG and testing potential therapies. For example, a single administration of the human NGLY1 gene to newborn rats (via intracerebroventricular injection) using an AAV9 delivery system resulted in increased NGLY1 activity in the brain and normalization of movement dysfunction52.
Human Cell Lines
Human cell-based disease models provide a resource to study disease pathophysiology and to develop treatments for NGLY1 deficiency.
Patient-derived fibroblasts
NGLY1 knockout fibroblast cell lines showed decreased NGLY1 expression, are resistant to hypotonic lysis increased sensitivity to mitochondrial inhibition and abnormal mitochondrial physiology22,34,35. Patient fibroblasts show much lower NGLY1 expression than other studies cell lines54.
Induced-pluripotent stem cell (iPSC) lines
Several iPS cell lines have been created. The NCATs-CL907 line was derived from dermal fibroblasts from a patient carrying a heterozygous mutation of NGLY1 (p.R390P and p.L319P)55 and the TRNDi002-B line was derived from a patient with a different heterozygous mutation (p.Q208X and p.G310G)56. The TRNDi010-C cell line was derived from a patient carrying a homozygous mutation (p.R401X)57.
Two gene corrected iPSC lines have been generated using a patient derived iPSC line (NCATS-CL6103). Using CRISPR/Cas9, the lines contain corrected alleles of NGLY1 (NCATS-CL6104) or (NCATS-CL6105)58.
Other cell lines
Numerous NGLY1 deficient human cell lines have been generated and studied. In general, NGLY1 expression varies significantly between different cell types which leads to variable dysregulation of other genes; a comparison of four different cell types (fibroblast, lymphoblastoid, neuronal progenitor, and fibroblast-derived induced pluripotent stem cells) from NGLY1-CDDG patients was performed and the data collated into a website54. However, downregulation of proteasomal genes is universal across all studied cell types (including models using immortalized cancer cells), which is consistent with NGLY1 as a regulator of the NRF1 transcription factor54,59.
Cerebral Organoids
In order to study the effect of NGLY1 deficiency on neural development in human tissue, cerebral organoids were prepared from human embryonic stem cells and induced pluripotent stem cells lacking normal NGLY1; the embryonic stem cells were edited using CRISPR-Cas9 to lack the gene and the induced pluripotent stem cells were derived from fibroblasts taken from NGLY1-CDDG patients60. Cerebral organoids prepared from these cells showed abnormal differentiation, defective formation of upper-layer neurons, and impairments to the signalling pathways necessary to sustain radial glial. In cerebral organoids lacking normal NGLY1 multiple transcription factors were also downregulated and the organoids were more sensitive to various stresses. These organoids may be used to study the effect of NGLY1 deficiency on the developing human brain.
Clinical Studies
Active
A Study of GlcNAc on Tear Production in NGLY1-CDDG (NCT0540245)
NGLY1-CDDG can lead to eye damage due to the inability to produce enough tears. A multi-center randomized, double-blind, placebo-controlled trial is being conducted to assess whether the dietary supplement, GlcNAc, improves tear production in NGLY1-CDDG.
The study will soon be recruiting participants. Families who are interested in participating can contact their nearest study site at the Children's Hospital of Philadelphia (youbim@chop.edu) or Seattle Children's Hospital (Hayden.Vreugdenhil@SeattleChildrens.org) by email.
Safety and Efficacy of GS-100 Gene Therapy in Patients With NGLY1 Deficiency (NCT06199531)
Grace Science, LLC is conducting a first in human open-label, dose escalation study designed to assess the safety and efficacy of a gene therapy for NGLY1 deficiency. An adeno-associated viral vector serotype 9 (AAV9) carrying the NGLY1 gene will be delivered via intracerebroventricular (ICV) injection to subjects diagnosed with NGLY1 deficiency. The study is currently recruiting participants. Individuals interested in participating in this study should contact gina@gracescience.com.
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorders of Glycosylation Consortium (FCDGC) is conducting a natural history study on all CDG types, including NGLY1-CDDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life. The published 5-year study results can be found here.
Completed
NGLY1 Deficiency: A Prospective Natural History Study (NCT03834987)
Stanford University and Grace Science Foundation completed a 32 month natural history study on NGYL1-CDDG with 29 participants. The purpose of this study is to understand the clinical spectrum and progression of NGLY1 deficiency, identify clinical biomarkers that can be used in therapeutic trials, and identify genotype-phenotype correlations. The published study results can be found here
Organizations
Grace Science Foundation
Grace Science LLC
Publications
NGLY1-CDDG Scientific Articles on PubMed
Additional Resources
Grace Science 'Guide to NGLY1'
NGLY1-CDDG GeneReviews
NGLY1 Deficiency on FCDGC
IEMbase
OMIM
Orphanet
GARD
NORD
Genetic Testing Registry
ClinVar
NIH
GeneCards
UniProt
References
- Enns, G. M. et al. Mutations in NGLY1 Cause an Inherited Disorder of the Endoplasmic Reticulum-Associated Degradation (ERAD) Pathway. Genet. Med. 16, 751–758 (2014).
- He, P. et al. A congenital disorder of deglycosylation: Biochemical characterization of N-glycanase 1 deficiency in patient fibroblasts. Glycobiology 25, 836–844 (2015).
- Suzuki, T. & Yoshida, Y. Ever-expanding NGLY1 biology. J. Biochem. 171, 141–143 (2022).
- Lipiński, P., Bogdańska, A., Różdżyńska-Świątkowska, A., Wierzbicka-Rucińska, A. & Tylki-Szymańska, A. NGLY1 deficiency: Novel patient, review of the literature and diagnostic algorithm. JIMD Rep. 51, 82–88 (2020).
- Lipari Pinto, P. et al. NGLY1 deficiency—A rare congenital disorder of deglycosylation. JIMD Rep. 53, 2–9 (2020).
- Rios-Flores, I. M. et al. Acute liver failure in a male patient with NGLY1-congenital disorder of deglycosylation. Eur. J. Med. Genet. 63, 103952 (2020).
- Lipiński, P., Cielecka-Kuszyk, J., Socha, P. & Tylki-Szymańska, A. Liver involvement in NGLY1 congenital disorder of deglycosylation. Polish J. Pathol. 71, 66–68 (2020).
- Ge, H. et al. Two novel compound heterozygous mutations in NGLY1as a cause of congenital disorder of deglycosylation: a case presentation. BMC Med. Genet. 21, 135 (2020).
- Kariminejad, A. et al. NGLY1 deficiency: Novel variants and literature review. Eur. J. Med. Genet. 64, 104146 (2021).
- Dabaj, I. et al. NGLY1 Deficiency: A Rare Newly Described Condition with a Typical Presentation. Life 11, 187 (2021).
- Lipiński, P., Bogdańska, A., Socha, P. & Tylki-Szymańska, A. Liver Involvement in Congenital Disorders of Glycosylation and Deglycosylation. Front. Pediatr. 9, 696918 (2021).
- Stuut, T., Popescu, O. & Oviedo, A. N-Glycanase 1 Deficiency Is a Rare Cause of Pediatric Neurodegeneration With Neuronal Inclusions and Liver Steatosis. Cureus 13, e19126 (2021).
- Kalfon, L. et al. Congenital Hypotonia: Cracking a SAGA of consanguineous kindred harboring four genetic variants. Mol. Genet. genomic Med. 10, e1849 (2022).
- Need, A. C. et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 49, 353–361 (2012).
- Levy, R. J., Frater, C. H., Gallentine, W. B., Phillips, J. M. & Ruzhnikov, M. R. Delineating the epilepsy phenotype of NGLY1 deficiency. J. Inherit. Metab. Dis. doi:10.1002/JIMD.12494.
- Pandey, A., Adams, J. M., Han, S. Y. & Jafar-Nejad, H. NGLY1 Deficiency, a Congenital Disorder of Deglycosylation: From Disease Gene Function to Pathophysiology. Cells 11, 1155 (2022).
- Caglayan, A. O. et al. NGLY1 Mutation Causes Neuromotor Impairment, Intellectual Disability, and Neuropathy. Eur. J. Med. Genet. 58, 39–43 (2015).
- Heeley, J. & Shinawi, M. Multi-systemic involvement in NGLY1-related disorder caused by two novel mutations. Am. J. Med. Genet. Part A 167, 816–820 (2015).
- Lam, C. et al. Prospective Phenotyping of NGLY1-CDDG, the First Congenital Disorder of Deglycosylation. Genet. Med. 19, 168 (2017).
- van Keulen, B. J., Rotteveel, J. & Finken, M. J. J. Unexplained death in patients with NGLY1 mutations may be explained by adrenal insufficiency. Physiol. Rep. 7, e13979 (2019).
- Cahan, E. M. & Frick, S. L. Orthopaedic phenotyping of NGLY1 deficiency using an international, family-led disease registry. Orphanet J. Rare Dis. 14, (2019).
- Panneman, D. M. et al. Variants in NGLY1 lead to intellectual disability, myoclonus epilepsy, sensorimotor axonal polyneuropathy and mitochondrial dysfunction. Clin. Genet. 97, 556–566 (2020).
- Abuduxikuer, K., Zou, L., Wang, L., Chen, L. & Wang, J.-S. Novel NGLY1 gene variants in Chinese children with global developmental delay, microcephaly, hypotonia, hypertransaminasemia, alacrimia, and feeding difficulty. J. Hum. Genet. 65, 387–396 (2020).
- Suzuki, T., Huang, C. & Fujihira, H. The cytoplasmic peptide:N-glycanase (NGLY1) - Structure, expression and cellular functions. Gene 577, 1–7 (2016).
- Yoshida, Y. et al. Loss of peptide:N-glycanase causes proteasome dysfunction mediated by a sugar-recognizing ubiquitin ligase. Proc. Natl. Acad. Sci. U. S. A. 118, e2102902118 (2021).
- Tomlin, F. M. et al. Inhibition of NGLY1 Inactivates the Transcription Factor Nrf1 and Potentiates Proteasome Inhibitor Cytotoxicity. ACS Cent. Sci. 3, 1143–1155 (2017).
- Yang, K., Huang, R., Fujihira, H., Suzuki, T. & Yan, N. N-glycanase NGLY1 regulates mitochondrial homeostasis and inflammation through NRF1. J. Exp. Med 215, 2600–2616 (2018).
- Suzuki, T. et al. Endo-β-N-acetylglucosaminidase forms N-GlcNAc protein aggregates during ER-associated degradation in Ngly1-defective cells. Proc. Natl. Acad. Sci. U. S. A. 112, 1398–1403 (2015).
- Forcina, G. C. et al. Ferroptosis regulation by the NGLY1/NFE2L1 pathway. Proc. Natl. Acad. Sci. U. S. A. 119, (2022).
- Galeone, A. et al. Regulation of BMP4/Dpp retrotranslocation and signaling by deglycosylation. Elife 9, 1–32 (2020).
- Galeone, A. et al. Tissue-specific regulation of BMP signaling by Drosophila N-glycanase 1. Elife 6, (2017).
- Talsness, D. M. et al. A Drosophila screen identifies NKCC1 as a modifier of NGLY1 deficiency. Elife 9, 1–22 (2020).
- Han, S. Y. et al. A conserved role for AMP-activated protein kinase in NGLY1 deficiency. PLoS Genet. 16, (2020).
- Tambe, M. A., Ng, B. G. & Freeze, H. H. N-Glycanase 1 Transcriptionally Regulates Aquaporins Independent of Its Enzymatic Activity. Cell Rep. 29, 4620-4631.e4 (2019).
- Kong, J. et al. Mitochondrial function requires NGLY1. Mitochondrion 38, 6–16 (2018).
- Wu, J. & Chen, Z. J. Innate Immune Sensing and Signaling of Cytosolic Nucleic Acids. Annu. Rev. Immunol. 32, 461–488 (2014).
- Fujihira, H. et al. Liver-specific deletion of Ngly1 causes abnormal nuclear morphology and lipid metabolism under food stress. Biochim. Biophys. acta. Mol. basis Dis. 1866, 165588 (2020).
- Lam, C., Wolfe, L., Need, A., Shashi, V. & Enns, G. NGLY1-Related Congenital Disorder of Deglycosylation. GeneReviews® (2018).
- Haijes, H. A. et al. Aspartylglycosamine is a biomarker for NGLY1-CDDG, a congenital disorder of deglycosylation. Mol. Genet. Metab. 127, 368–372 (2019).
- Mueller, W. F. et al. GlcNAc-Asn is a biomarker for NGLY1 deficiency. J. Biochem. 171, 177–186 (2022).
- Hall, P. L. et al. Urine oligosaccharide screening by MALDI-TOF for the identification of NGLY1 deficiency. Mol. Genet. Metab. 124, 82–86 (2018).
- Iyer, S. et al. Drug screens of NGLY1 deficiency in worm and fly models reveal catecholamine, NRF2 and anti-inflammatory-pathway activation as potential clinical approaches. Dis. Model. Mech. 12, (2019).
- Bi, Y., Might, M., Vankayalapati, H. & Kuberan, B. Repurposing of Proton Pump Inhibitors as First Identified Small Molecule Inhibitors of Endo-β-N-acetylglucosaminidase (ENGase) for the Treatment of Rare NGLY1 Genetic Disease. Bioorg. Med. Chem. Lett. 27, 2962 (2017).
- Fujihira, H., Asahina, M. & Suzuki, T. Physiological importance of NGLY1, as revealed by rodent model analyses. J. Biochem. 171, 161–167 (2022).
- Lehrbach, N. J. NGLY1: insights from Caenorhabditis elegans. J. Biochem. 171, 145–152 (2022).
- Pandey, A. & Jafar-Nejad, H. Tracing the NGLY1 footprints: insights from Drosophila. J. Biochem. 171, 153–160 (2022).
- Lehrbach, N. J. & Ruvkun, G. Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. Elife 5, e17721 (2016).
- Owings, K. G., Lowry, J. B., Bi, Y., Might, M. & Chow, C. Y. Transcriptome and functional analysis in a Drosophila model of NGLY1 deficiency provides insight into therapeutic approaches. Hum. Mol. Genet. 27, 1055–1066 (2018).
- Rodriguez, T. P. et al. Defects in the Neuroendocrine Axis Contribute to Global Development Delay in a Drosophila Model of NGLY1 Deficiency. G3 (Bethesda). 8, 2193–2204 (2018).
- Fujihira, H. et al. Lethality of mice bearing a knockout of the Ngly1-gene is partially rescued by the additional deletion of the Engase gene. PLoS Genet. 13, (2017).
- Asahina, M. et al. JF1/B6F1 Ngly1 -/- mouse as an isogenic animal model of NGLY1 deficiency. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 97, 89–102 (2021).
- Asahina, M. et al. Reversibility of motor dysfunction in the rat model of NGLY1 deficiency. Mol. Brain 14, (2021).
- Asahina, M. et al. Ngly1 -/- rats develop neurodegenerative phenotypes and pathological abnormalities in their peripheral and central nervous systems. Hum. Mol. Genet. 29, 1635–1647 (2020).
- Rauscher, B. et al. Patient-derived gene and protein expression signatures of NGLY1 deficiency. J. Biochem. 171, 187–199 (2022).
- Pradhan, M. et al. An induced pluripotent stem cell line (NCATS-CL9075) from a patient carrying compound heterozygote mutations, p.R390P and p.L318P, in the NGLY1 gene. Stem Cell Res. 54, (2021).
- Li, R. et al. Generation of an induced pluripotent stem cell line (TRNDi002-B) from a patient carrying compound heterozygous p.Q208X and p.G310G mutations in the NGLY1 gene. Stem Cell Res. 34, (2019).
- Yang, S. et al. An induced pluripotent stem cell line (TRNDi010-C) from a patient carrying a homozygous p.R401X mutation in the NGLY1 gene. Stem Cell Res. 39, (2019).
- Pavlinov, I. et al. Generation of two gene corrected human isogenic iPSC lines (NCATS-CL6104 and NCATS-CL6105) from a patient line (NCATS-CL6103) carrying a homozygous p.R401X mutation in the NGLY1 gene using CRISPR/Cas9. Stem Cell Res. 56, (2021).
- Mueller, W. F. et al. Loss of N-Glycanase 1 Alters Transcriptional and Translational Regulation in K562 Cell Lines. G3 (Bethesda). 10, 1585–1597 (2020).
- Lin, V. J. T. et al. Deficiency of N-glycanase 1 perturbs neurogenesis and cerebral development modeled by human organoids. Cell Death Dis. 13, (2022).
- Zhu, L. et al. AAV9-NGLY1 gene replacement therapy improves phenotypic and biomarker endpoints in a rat model of NGLY1 Deficiency.
Mol Ther Methods Clin Dev. 27, (2022).