
Lay Summary
MPI-CDG, formerly known as CDG-Ib, is a rare but treatable inherited condition that mainly affects the liver and gastrointestinal system. To date, 35 cases of MPI-CDG have been reported in the literature. MPI-CDG is classified as a disorder of N-linked protein glycosylation. MPI-CDG is caused when an individual has mutations in both copies of the MPI gene which provides instructions for making an enzyme that generates the sugar mannose-6-phosphate. Mannose-6 phosphate is required for generating molecules that donate mannose sugars onto growing sugar chains during N-glycosylation. Mutations in the MPI gene cause some proteins to have incomplete or absent sugar chains. Individuals affected by MPI-CDG can present a range of symptoms, but common clinical features include liver and gastrointestinal abnormalities, blood clotting defects, and low blood glucose levels; these symptoms typically present in infancy. Several screening tests are available for MPI-CDG, but a definitive diagnosis is achieved through genetic testing. Early diagnosis is crucial for MPI-CDG, as it is a potentially life-threatening disease, but is treatable through dietary supplements of mannose.
Overview
Mannose phosphate isomerase congenital disorder of glycosylation (MPI-CDG) is a rare autosomal recessive disorder that arises from defects in the MPI gene. MPI encodes the enzyme: mannose phosphate isomerase (MPI)1. MPI converts fructose-6-phosphate to mannose-6-phosphate, which is needed for N-glycosylation2. Mannose-6-phosphate is necessary for the generation of GDP-mannose and dolichol-phosphate-mannose: the mannose donors used in the construction of the lipid-linked oligosaccharide during N-glycosylation. Deficiency in MPI results in insufficient N-glycosylation of proteins.
MPI-CDG was first clinically reported in 19803, but it was not until 1998 that genetic mutations in MPI were first described4,5,6. Collectively, 40 MPI-CDG cases have been reported in the literature, with 28 cases that have been confirmed by genetic testing1–30. Onset of symptoms can present from infancy, where common clinical features include liver, gastrointestinal and blood clotting abnormalities—although symptoms and severity vary between affected individuals1. MPI-CDG are typically diagnosed through a combination of biochemical (transferrin analysis), enzymatic (assessment of MPI), and genetic testing. As MPI affects N-glycosylation, transferrin isoform analysis is usually performed during diagnosis, with cases presenting abnormal transferrin isoform patterns (typically type I)30. However, genetic testing typically provides a definitive diagnosis. MPI-CDG can be treated with mannose therapy, which involves dietary supplements of the monosaccharide: mannose.
Synonyms
- Mannose phosphate isomerase-congenital disorder of glycosylation
- Mannose phosphate isomerase deficiency-CDG
- Phosphomannose isomerase deficiency
- CDG type 1b
- CDGIb
- CDG1B
- Protein-losing enteropathy-hepatic fibrosis syndrome
- Saguenay-Lac Saint-Jean syndrome
Inheritance
MPI-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The MPI gene encodes mannose phosphate isomerase (MPI), also known as phosphomannose isomerase. Isomerase enzymes alter the structure of molecules by rearranging their atoms, generating a different molecule (or isomer)31. MPI is typically found in the cytosol, where it catalyzes the conversion of fructose-6-phsophate to mannose-6-phosphate30.
Mannose-6-phosphate is needed to generate mannose-1-phosphate, which a precursor of the nucleotide sugar GDP-mannose32. Nucleotide sugars are also called glycosyl donors or sugar donors because they enable sugars to be added onto other molecules. The generation of mannose containing glycosyl donors is particularly important for N-glycosylation of proteins, which involves the attachment of a mannose-rich glycan to the amino acid asparagine on a protein33.
Monosaccharide Interconversion
The monosaccharide mannose is an important component of glycans that are attached to various proteins and lipids. Before mannose can be incorporated into glycans, it must be converted to GDP-mannose in the cytoplasm (Figure 1) 34. Several steps are required to generate GDP-mannose from mannose, which includes the generation of mannose-6-phosphate. Mannose-6-phosphate can be generated by two ways: 1) by MPI converting fructose-6-phosphate to mannose-6-phosphate or 2) by an enzyme (hexokinase) adding phosphate to mannose forming mannose-6-phosphate32. Once generated, the PMM2 enzyme converts mannose-6-phosphate to mannose-1-phosphate, which is used to synthesize GDP-mannose, the mannose donor in the cytoplasm. GDP-mannose is also a precursor for dolichol-P-mannose, the mannose donor used inside the ER35.
The first step of N-glycosylation is the building of a mannose-rich lipid-linked oligosaccharide (LLO) comprised of a 14-sugar oligosaccharide and dolichol pyrophosphate (Dol-PP). The oligosaccharide component, also referred to as the oligosaccharide precursor, is subsequently transferred en bloc to an asparagine (Asn; N) residue of a protein inside the ER. The synthesis of the LLO occurs through a stepwise process that occurs both in the cytoplasm and inside the ER, and thus requires both GDP-mannose and Dol-P-mannose as glycosyl donors33.
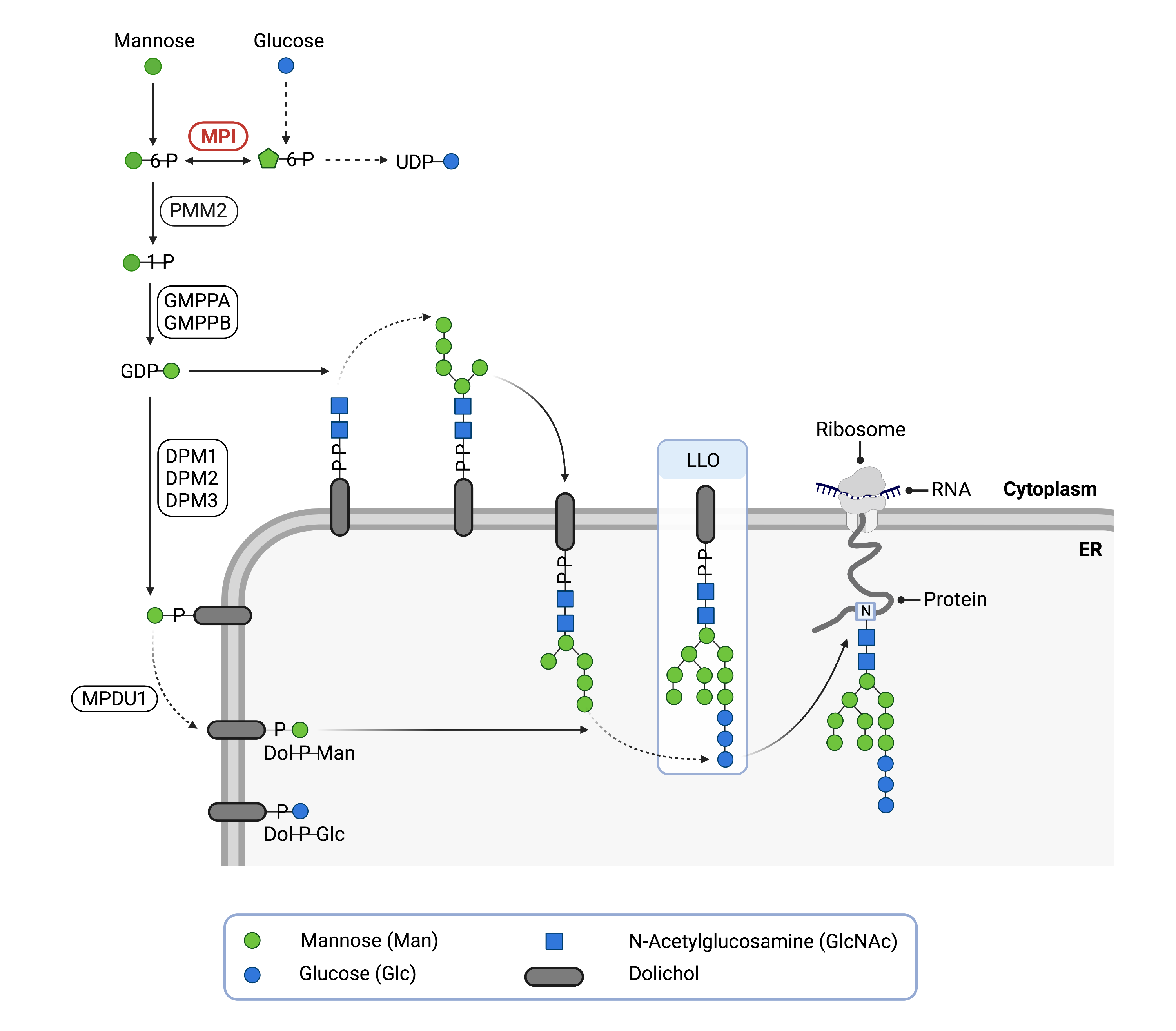
Figure 1. Role of MPI in glycosylation.
MPI converts fructose-6-phosphate into mannose-6-phosphate, which is needed for N-glycosylation. Mannose-6-phosphate is needed for cells to produce activated forms of mannose (GDP-mannose and Dol-P-mannose), which are used to generate the lipid-linked oligosaccharide (LLO) during N-glycosylation, as well as N-glycan chain elongation.
Disease Mechanism
Defects in MPI reduce the ability of MPI to generate mannose-6-phsophate, which leads to abnormal N-glycans2. Deficient MPI are unable or at a reduced capacity of converting fructose-6-phohsphate to mannose-6-phosphate. Fructose-6-phosphate can be used in a different pathway (glycolytic pathway), reducing accumulation in cells, which may limit neurological effects in individuals with MPI-CDG36. However, reduced activity of MPI by gene mutations can lead to abnormal N-glycans structures and levels on proteins30. As mannose-6-phosphate can also be generated through a separate enzymatic pathway, providing cells with more mannose (such as by mannose dietary supplements) can increase levels of GDP-mannose, generating normal N-glycans and alleviating symptoms37.
Mutations
To date, 21 pathogenic variants have been identified in the MPI gene, with the most common variant detected being p.Arg219Gln (c.656G > A). Of these pathogen variants, 17 are missense variants, 2 are frameshift variants, 1 is a nonsense variant, and 1 is a splice variant2,30. Individuals with MPI-CDG can also be found to have different mutations in each copy of the MPI gene (known as compound heterozygosity)5,9.
Some variants can also yield clinically asymptomatic MPI-CDG10. However, the extent of enzymatic activity does not appear to predict disease severity30.
Signs & Symptoms
Clinical Presentation
Affected individuals typically present clinical features from infancy, with disease characterised by liver and gastrointestinal abnormalities. MPI-CDG symptoms can include:
- Gastrointestinal abnormalities – vomiting, diarrhoea, failure to thrive, excess loss of proteins to the intestine (protein-losing enteropathy)30,38
- Liver abnormalities – liver fibrosis, enlarged liver (hepatomegaly), liver disease30
In addition, while affected individuals are not typically associated with neurological symptoms, some individuals can have seizures30.
Biochemical Abnormalties
Individuals with MPI-CDG can often present with elevated liver enzymes (arylsulfatase A and transaminases), blood clotting (coagulation) abnormalities, low blood albumin (hypoalbuminemia) levels, and low blood glucose levels (hypoglycemia). In addition, affected individuals can also present reduced hormone levels (hypothyroidism)30.
Classification
MPI-CDG is classified as a disorder of N-linked protein glycosylation. More specifically, it is a disorder of monosaccharide interconversion.
Under the former CDG classification system, MPI-CDG is classified as a Type I CDG, which arise due to defects in the synthesis of N-glycoproteins that occur before the glycan is transferred from the LLO onto the protein.
Diagnosis
MPI should be suspected when individuals present liver and gastrointestinal symptoms, without displaying central nervous system defects20—although, minor neurological defects can also be found in affected individuals39. To date, common symptoms include diarrhoea, low blood sugar levels, failure to thrive, protein-losing enteropathy, liver pathologies and antithrombin deficiencies27.
Screening in suspected patients typically begins with a blood test to analyze serum transferrin isoforms. Direct assessment of MPI enzymatic activity may also be used and is often performed after a transferrin analysis displays a type I pattern. Genetic testing is used to definitively diagnose MPI-CDG30.
Transferrin Analysis
Individuals with MPI-CDG show a characteristic type I pattern by transferrin isoelectric focusing (TIEF) or mass spectrometry analysis of transferrin30. Type I patterns are observed in CDG that arise due to defects in LLO assembly and are characterized by a decrease in tetrasialo-transferrin and an increase in di-sialo and a-sialo transferrin isoforms30.
MPI Enzymatic Acitivity
Defects in MPI enzyme activity can be assessed by enzyme analysis. Typically, enzyme analysis is performed on patient fibroblasts or leukocytes30.
Biomarkers
Several biomarkers have been identified for MPI-CDG which may aid in diagnosis.
N-Tetrasaccharide (Neu5Acα2,6Galβ1,4-GlcNAcβ1,4GlcNA)
N-tetrasaccharide (Neu5Acα2,6Galβ1,4-GlcNAcβ1,4GlcNA) has been found at higher levels in plasma from patients with MPI-CDG compared to unaffected controls, with N-tetrasaccharide levels reducing after mannose treatment. This N-tetrasaccharide was also found at higher levels in patients with ALG1-CDG and PMM2-CDG40.
Aspartylglucosaminidase
Increased levels of plasma aspartylglucosaminidase activity have been found in many different types of CDG, including MPI-CDG41.
Intercellular Adhesion Molecule 1 (ICAM-1)
Reduced intercellular adhesion molecule 1 (ICAM-1) protein levels have been found at the cell surface of fibroblasts from MPI-CDG patients, compared to controls. ICAM-1 levels increased after mannose treatment. ICAM-1 protein levels were also found reduced in other CDG, including PMM2-CDG and ALG1-CDG42.
Prognosis
MPI-CDG prognosis can vary among affected individuals, depending on disease severity. Some affected individuals can be asymptomatic in adulthood, with one individual reported in the medical literature in 2014 as being asymptomatic at 32 years old10. MPI-CDG symptoms can be effectively managed in individuals with mannose therapy4,6,16,18. However, MPI-CDG can be life-threatening, with premature death observed in untreated individuals presenting symptoms in early life20. Death can be linked to liver failure and sepsis in some affected individuals30. Liver abnormalities can persist even with mannose therapy, and poor compliance to mannose therapy can lead to MPI-CDG symptoms recurring (such as diarrhea and thrombosis)37.
Management
MPI-CDG may involve gastrointestinal management (such as albumin infusions or intravenous feeding), endocrine management (such as frequent feeding or intravenous glucose) liver transplants, mannose therapy, and blood clotting management. Affected individuals may also regularly undergo tests to monitor liver, gastrointestinal, endocrine, haematological, neurological, kidney and immune function, as well as mannose therapy monitoring30.
Therapies
In addition to managing symptoms, mannose therapy (mannose supplements) can be used to treat MPI-CDG. Mannose supplements can alleviate symptoms, improving N-glycosylation of proteins. This is achieved by providing more mannose for hexokinase to generate mannose-6-phosphate for GDP-mannose synthesis. As mannose therapy can have limited results on liver disease, liver transplants are another therapeutic option21,24.
Research Models
Several research models have been developed for studying MPI, including zebrafish, mice, and human cell lines43.
Fish (D. rerio)
Zebrafish models with reduced levels of MPI displayed decreased LLO and N-glycan levels, 50% embryonic lethality levels (by four days after fertilization), and abnormal features in the majority of the larvae that survived. However, normal phenotypes were obtained if mannose was added to the water within 24 hours after fertilization44.
Mouse (M. musculus)
Mpi-/- constitutive knockout mice
Mpi-/- knockout mice models displayed embryonic lethal phenotypes, as well as growth defects, which was linked to toxic accumulation of mannose-6-phosphate (studies included Mpi-/- primary embryonic fibroblasts)32.
Mpi hypomorphic mice
Mpi hypomorphic mice displayed roughly 15% embryonic lethality. Embryonic lethality was increased when mannose supplements were administered during pregnancy, and around half of the surviving pups developed eye defects45.
Human Cell Lines
Human colon adenocarcinoma cells (HT-29 cell line)
HT-29 cells were used and MPI levels were reduced using siRNAs. By labelling mannose, experiments showed that mannose was redirected to use by a separate enzyme (PMM2) when cells lacked MPI, increasing mannose-modified protein levels46.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including MPI-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Unknown
Study of ORL-1M (D-mannose) in Patients With CDG-Ib (NCT03404869)
Orpha Labs is conducting an interventional study (clinical trial) on MPI-CDG. The purpose of this study is to assess ability of mannose to treat hypoglycemia, diarrhea and vomiting and improve serum transferrin glycosylation patterns in MPI-CDG patients.
Publications
MPI-CDG Scientific Articles on PubMed
Consensus guideline for the diagnosis and management of mannose phosphate isomerase-congenital disorder of glycosylation
Additional Resources
MPI-CDG on FCDGC
MPI-CDG Infographic
MPI-CDG Clinical Utility Gene Card
IEMbase
OMIM
OrphaNet
GARD
Genetic Testing Registry
ClinVar
NIH
GeneCards
UniProt
References
- Mühlhausen C, Henneke L, Schlotawa L, et al. Mannose phosphate isomerase deficiency‐congenital disorder of glycosylation ( <scp>MPI‐CDG</scp> ) with cerebral venous sinus thrombosis as first and only presenting symptom: A rare but treatable cause of thrombophilia. JIMD Reports. 2020;55(1). doi:10.1002/jmd2.12149
- Abdel Ghaffar TY, Ng BG, Elsayed SM, et al. <scp>MPI‐CDG</scp> from a hepatic perspective: Report of two Egyptian cases and review of literature. JIMD Reports. 2020;56(1). doi:10.1002/jmd2.12159
- PEDERSEN PS, TYGSTRUP I. CONGENITAL HEPATIC FIBROSIS COMBINED WITH PROTEIN-LOSING ENTEROPATHY AND RECURRENT THROMBOSIS. Acta Paediatrica. 1980;69(4). doi:10.1111/j.1651-2227.1980.tb07136.x
- Niehues R, Hasilik M, Alton G, et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. Journal of Clinical Investigation. 1998;101(7). doi:10.1172/JCI2350
- Jaeken J, Matthijs G, Saudubray JM, et al. Phosphomannose Isomerase Deficiency: A Carbohydrate-Deficient Glycoprotein Syndrome with Hepatic-Intestinal Presentation. The American Journal of Human Genetics. 1998;62(6). doi:10.1086/301873
- Westphal V, Kjaergaard S, Davis JA, Peterson SM, Skovby F, Freeze HH. Genetic and Metabolic Analysis of the First Adult with Congenital Disorder of Glycosylation Type Ib: Long-Term Outcome and Effects of Mannose Supplementation. Molecular Genetics and Metabolism. 2001;73(1). doi:10.1006/mgme.2001.3161
- Pelletier VA, Galéano N, Brochu P, Morin CL, Weber AM, Roy CC. Secretory diarrhea with protein-losing enteropathy, enterocolitis cystica superficialis, intestinal lymphangiectasia, and congenital hepatic fibrosis: A new syndrome. The Journal of Pediatrics. 1986;108(1). doi:10.1016/S0022-3476(86)80769-7
- Vuillaumier-Barrot S. Protein losing enteropathy-hepatic fibrosis syndrome in Saguenay-Lac St-Jean, Quebec is a congenital disorder of glycosylation type Ib. Journal of Medical Genetics. 2002;39(11). doi:10.1136/jmg.39.11.849
- Schollen E, Dorland L, de Koning TJ, et al. Genomic organization of the human phosphomannose isomerase (MPI) gene and mutation analysis in patients with congenital disorders of glycosylation type Ib (CDG-Ib). Human Mutation. 2000;16(3). doi:10.1002/1098-1004(200009)16:3<247::AID-HUMU7>3.0.CO;2-A
- Helander A, Jaeken J, Matthijs G, Eggertsen G. Asymptomatic phosphomannose isomerase deficiency (MPI-CDG) initially mistaken for excessive alcohol consumption. Clinica Chimica Acta. 2014;431. doi:10.1016/j.cca.2014.01.018
- de Koning TJ, Dorland L, van Diggelen OP, et al. A Novel Disorder of N-Glycosylation Due to Phosphomannose Isomerase Deficiency. Biochemical and Biophysical Research Communications. 1998;245(1). doi:10.1006/bbrc.1998.8385
- de Koning TJ, Nikkels PGJ, Dorland L, et al. Congenital hepatic fibrosis in 3 siblings with phosphomannose isomerase deficiency. Virchows Archiv. 2000;437(1). doi:10.1007/s004280000185
- de Lonlay P, Cuer M, Vuillaumier-Barrot S, et al. Hyperinsulinemic hypoglycemia as a presenting sign in phosphomannose isomerase deficiency: A new manifestation of carbohydrate-deficient glycoprotein syndrome treatable with mannose. The Journal of Pediatrics. 1999;135(3). doi:10.1016/S0022-3476(99)70139-3
- Penel-Capelle D, Dobbelaere D, Jaeken J, Klein A, Cartigny M. Congenital disorder of glycosylation Ib (CDG-Ib) without gastrointestinal symptoms. Journal of Inherited Metabolic Disease. 2003;26(1). doi:10.1023/A:1024044017385
- Damen G, de Klerk H, Huijmans J, den Hollander J, Sinaasappel M. Gastrointestinal and Other Clinical Manifestations in 17 Children With Congenital Disorders of Glycosylation Type Ia, Ib, and Ic. Journal of Pediatric Gastroenterology and Nutrition. 2004;38(3). doi:10.1097/00005176-200403000-00010
- Harms HK, Zimmer KP, Kurnik K, Bertele-Harms R, Weidinger S, Reiter K. Oral mannose therapy persistently corrects the severe clinical symptoms and biochemical abnormalities of phosphomannose isomerase deficiency. Acta Paediatrica. 2002;91(10). doi:10.1080/080352502760311566
- Babovic-Vuksanovic D, Patterson MC, Schwenk WF, et al. Severe hypoglycemia as a presenting symptom of carbohydrate-deficient glycoprotein syndrome. The Journal of Pediatrics. 1999;135(6). doi:10.1016/S0022-3476(99)70103-4
- Hendriksz CJ, McClean P, Henderson M J, et al. Successful treatment of carbohydrate deficient glycoprotein syndrome type 1b with oral mannose. Archives of Disease in Childhood. 2001;85(4):339-340. doi:10.1136/adc.85.4.339
- de Lonlay P, Seta N. The clinical spectrum of phosphomannose isomerase deficiency, with an evaluation of mannose treatment for CDG-Ib. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2009;1792(9). doi:10.1016/j.bbadis.2008.11.012
- Vuillaumier-Barrot S. Protein losing enteropathy-hepatic fibrosis syndrome in Saguenay-Lac St-Jean, Quebec is a congenital disorder of glycosylation type Ib. Journal of Medical Genetics. 2002;39(11). doi:10.1136/jmg.39.11.849
- Mention K, Lacaille F, Valayannopoulos V, et al. Development of liver disease despite mannose treatment in two patients with CDG-Ib. Molecular Genetics and Metabolism. 2008;93(1). doi:10.1016/j.ymgme.2007.08.126
- Deeb A, al Amoodi A. A novel homozygous mutation in the mannose phosphate isomerase gene causing congenital disorder of glycation and hyperinsulinemic hypoglycemia in an infant. Clinical Case Reports. 2018;6(3). doi:10.1002/ccr3.1387
- de Lonlay P, Seta N, Barrot S, et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. Journal of Medical Genetics. 2001;38(1). doi:10.1136/jmg.38.1.14
- Janssen MCH, de Kleine RH, van den Berg AP, et al. Successful Liver Transplantation and Long-Term Follow-up in a Patient With MPI-CDG. PEDIATRICS. 2014;134(1). doi:10.1542/peds.2013-2732
- Martín Hernández E, Vega Pajares AI, Pérez González B, et al. Defecto congénito de glucosilación tipo Ib. Experiencia en el tratamiento con manosa. Anales de Pediatría. 2008;69(4). doi:10.1157/13126562
- Kelly D, Boneh A, Pitsch S, et al. Carbohydrate-deficient glycoprotein syndrome 1b: A new answer to an old diagnostic dilemma. Journal of Paediatrics and Child Health. 2001;37(5). doi:10.1046/j.1440-1754.2001.00671.x
- de la Morena-Barrio ME, Wypasek E, Owczarek D, et al. MPI-CDG with transient hypoglycosylation and antithrombin deficiency. Haematologica. 2019;104(2). doi:10.3324/haematol.2018.211326
- Tamminga RYJ, Lefeber DJ, Kamps WA, van Spronsen FJ. RECURRENT THROMBO-EMBOLISM IN A CHILD WITH A CONGENITAL DISORDER OF GLYCOSYLATION (CDG) TYPE IB AND TREATMENT WITH MANNOSE. Pediatric Hematology and Oncology. 2008;25(8). doi:10.1080/08880010802394616
- Liem YS, Bode L, Freeze HH, Leebeek FW, Zandbergen AA, Paul Wilson J. Using heparin therapy to reverse protein-losing enteropathy in a patient with CDG-Ib. Nature Clinical Practice Gastroenterology & Hepatology. 2008;5(4). doi:10.1038/ncpgasthep1061
- Čechová A, Altassan R, Borgel D, et al. Consensus guideline for the diagnosis and management of mannose phosphate isomerase‐congenital disorder of glycosylation. Journal of Inherited Metabolic Disease. 2020;43(4). doi:10.1002/jimd.12241
- Martinez Cuesta S, Furnham N, Rahman SA, Sillitoe I, Thornton JM. The evolution of enzyme function in the isomerases. Current Opinion in Structural Biology. 2014;26. doi:10.1016/j.sbi.2014.06.002
- DeRossi C, Bode L, Eklund EA, et al. Ablation of mouse phosphomannose isomerase (Mpi) causes mannose 6-phosphate accumulation, toxicity, and embryonic lethality. Journal of Biological Chemistry. 2006;281(9). doi:10.1074/jbc.M511982200
- Aebi M. N-linked protein glycosylation in the ER. Biochimica et Biophysica Acta - Molecular Cell Research. 2013;1833(11). doi:10.1016/j.bbamcr.2013.04.001
- Banerjee DK, Zhang Z, Baksi K, Serrano-Negrón JE. Dolichol phosphate mannose synthase: a Glycosyltransferase with Unity in molecular diversities. Glycoconjugate Journal. 2017;34(4). doi:10.1007/s10719-017-9777-4
- Freeze HH. Towards a therapy for phosphomannomutase 2 deficiency, the defect in CDG-Ia patients. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2009;1792(9). doi:10.1016/j.bbadis.2009.01.004
- Chang IJ, He M, Lam CT. Congenital disorders of glycosylation. Annals of Translational Medicine. 2018;6(24). doi:10.21037/atm.2018.10.45
- Girard M, Douillard C, Debray D, et al. Long term outcome of MPI-CDG patients on D-mannose therapy. Journal of Inherited Metabolic Disease. 2020;43(6). doi:10.1002/jimd.12289
- Levitt D, Levitt M. Protein losing enteropathy: comprehensive review of the mechanistic association with clinical and subclinical disease states. Clinical and Experimental Gastroenterology. 2017;Volume 10. doi:10.2147/CEG.S136803
- Jaeken J. Congenital disorders of glycosylation. Annals of the New York Academy of Sciences. 2010;1214(1):190-198. doi:10.1111/j.1749-6632.2010.05840.x
- Zhang W, James PM, Ng BG, et al. A novel N-tetrasaccharide in patients with congenital disorders of glycosylation, including asparagine-linked glycosylation protein 1, phosphomannomutase 2, and mannose phosphate isomerase deficiencies. Clinical Chemistry. 2016;62(1). doi:10.1373/clinchem.2015.243279
- Jackson M, Clayton P, Grunewald S, et al. Elevation of plasma aspartylglucosaminidase is a useful marker for the congenital disorders of glycosylation type I (CDG I). Journal of Inherited Metabolic Disease. 2005;28(6). doi:10.1007/s10545-005-0157-z
- He P, Ng BG, Losfeld ME, Zhu W, Freeze HH. Identification of intercellular cell adhesion molecule 1 (ICAM-1) as a hypoglycosylation marker in congenital disorders of glycosylation cells. Journal of Biological Chemistry. 2012;287(22). doi:10.1074/jbc.M112.355677
- Brasil S, Pascoal C, Francisco R, et al. CDG Therapies: From Bench to Bedside. International Journal of Molecular Sciences. 2018;19(5). doi:10.3390/ijms19051304
- Chu J, Mir A, Gao N, et al. A zebrafish model of congenital disorders of glycosylation with phosphomannose isomerase deficiency reveals an early opportunity for corrective mannose supplementation. Disease Models & Mechanisms. Published online January 1, 2012. doi:10.1242/dmm.010116
- Sharma V, Nayak J, DeRossi C, et al. Mannose supplements induce embryonic lethality and blindness in phosphomannose isomerase hypomorphic mice. FASEB Journal. 2014;28(4). doi:10.1096/fj.13-245514
- Sharma V, Ichikawa M, He P, et al. Phosphomannose Isomerase Inhibitors Improve N-Glycosylation in Selected Phosphomannomutase-deficient Fibroblasts. Journal of Biological Chemistry. 2011;286(45). doi:10.1074/jbc.M111.285502