
Lay Summary
GPAA1-CDG, also known as Glycosylphosphatidylinositol biosynthesis defect 15 (GPIBD15), is a rare inherited condition that affects multiple systems in the body. To date, fewer than 30 cases have been reported in the medical literature. GPAA1-CDG is classified as a disorder of GPI anchor biosynthesis and is caused when an individual has mutations in both copies of the GPAA1 gene. This gene provides instructions for making a protein involved in the attachment of a GPI anchor to a protein. GPI anchors are molecules that attach proteins to the surface of the cell. The GPAA1 protein is part of a group of enzymes that function together to attach GPI anchors to newly synthesized proteins, forming a GPI-anchored protein. Mutations in the GPAA1 gene impact this attachment step, resulting in GPI-anchored proteins which are unstable or unable to attach to the cell surface. Symptoms of GPAA1-CDG typically begin in early infancy or early childhood, and are primarily characterized by developmental delay, seizures, intellectual disability, shrinking of a brain region called the cerebellum (cerebellar atrophy), loss of bone density (osteopenia), low muscle tone (hypotonia), difficulties in muscle coordination (ataxia), abnormal eye movements (nystagmus), and distinctive facial features. Genetic testing is usually conducted to definitively diagnose GPAA1-CDG, which can be supported through biochemical analysis to measure levels of GPI-anchored proteins present on the surface of blood and skin cells. Brain imaging is often used to examine structural differences in the brain that may be associated with seizures or other neurological symptoms, helping to support diagnosis and guide care. There are currently no approved treatments for GPAA1-CDG, and treatments are largely focused on the management of specific symptoms and preventing disease complications.
Overview
Glycosylphosphatidylinositol Anchor Attachment 1 Congenital Disorder of Glycosylation (GPAA1-CDG) is a rare autosomal recessive genetic disorder resulting from biallelic variants in the GPAA1 gene. GPAA1-CDG is classified as a disorder of glycosylphosphatidylinositol (GPI) anchor biosynthesis. Fewer than 25 cases have been reported in the medical literature to date1–7, with the first 10 cases being reported in 20171.
The GPAA1 gene encodes a subunit of the GPI transamidase (GPI-TA) enzyme complex which facilitates the attachment of GPI anchors to proteins during GPI-anchored protein (GPI-AP) biosynthesis. Through this mechanism, GPI anchors are essential to the attachment of proteins to the cell surface, forming GPI-APs. The GPAA1 subunit catalyzes the formation of a peptide bond between the lipid component of a GPI anchor and a target protein8. Mutations in GPAA1 lead to deficiency in the levels of both GPI-TA and surface GPI-AP expression1,2,9.
Symptoms typically begin in early infancy or early childhood, and are characterized by developmental delay, intellectual disability, low muscle tone (hypotonia), loss of muscle coordination (including ataxia, dysmetria, and dysarthria), irregular eye movements (nystagmus), loss of bone density (osteopenia), and dysmorphic features1–4,6.
GPAA1-CDG is typically diagnosed through genetic testing1–7, which may be supported by biochemical tests including flow cytometry to examine GPI-AP expression on patient cells or measuring serum alkaline phosphatase (ALP) levels1–3,7. Additional evaluations often include neuroimaging (MRI) to examine brain structure abnormalities or EEG to detect seizure activity1–4. Currently, there is no cure or treatment for GPAA1-CDG, and therapy is based on management of symptoms, particularly seizures, and preventing disease complications.
Synonyms
- Glycosylphosphatidylinositol biosynthesis defect 15 (GPIBD15)
- Developmental delay, epilepsy, cerebellar atrophy, and osteopenia
- Neurodevelopmental delay-seizures-ophthalmic-anomalies-osteopenia-cerebellar atrophy syndrome
Inheritance
GPAA1-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The GPAA1 gene encodes the GPAA1 subunit of the GPI-TA complex, which is comprised of five subunits and catalyzes the attachment of a GPI anchor to a target protein to form a GPI-AP. GPAA1 carries out the final step in this protein attachment process by catalyzing the formation of a peptide bond between the lipid component of a GPI anchor to the C-terminal of the target protein. This process takes place in the lumen of the endoplasmic reticulum (ER). GPAA1 is a transmembrane protein8.
GPI-ANCHORED PROTEIN BIOSYNTHESIS
GPI-anchored protein biosynthesis is one of the major glycosylation pathways that attach glycans to lipid molecules within cells. Many proteins, called GPI-anchored proteins, are attached to the cell surface by GPI-anchors.
The mature GPI core structure consists of phosphatidylinositol (PI), glucosamine (GlcN), three mannose sugars (Man3), and two phosphoethanolamine groups (EtN-P2) (Figure 1). GPI-anchored proteins are attached to the GPI by forming a bond between the EtN-P group of the GPI core and the C-terminus of newly synthesized proteins in the ER lumen. In some GPI-APS, the core glycan is modified with a fourth mannose and/or N-acetylglucosamine (Glc-NAc) side chains8.
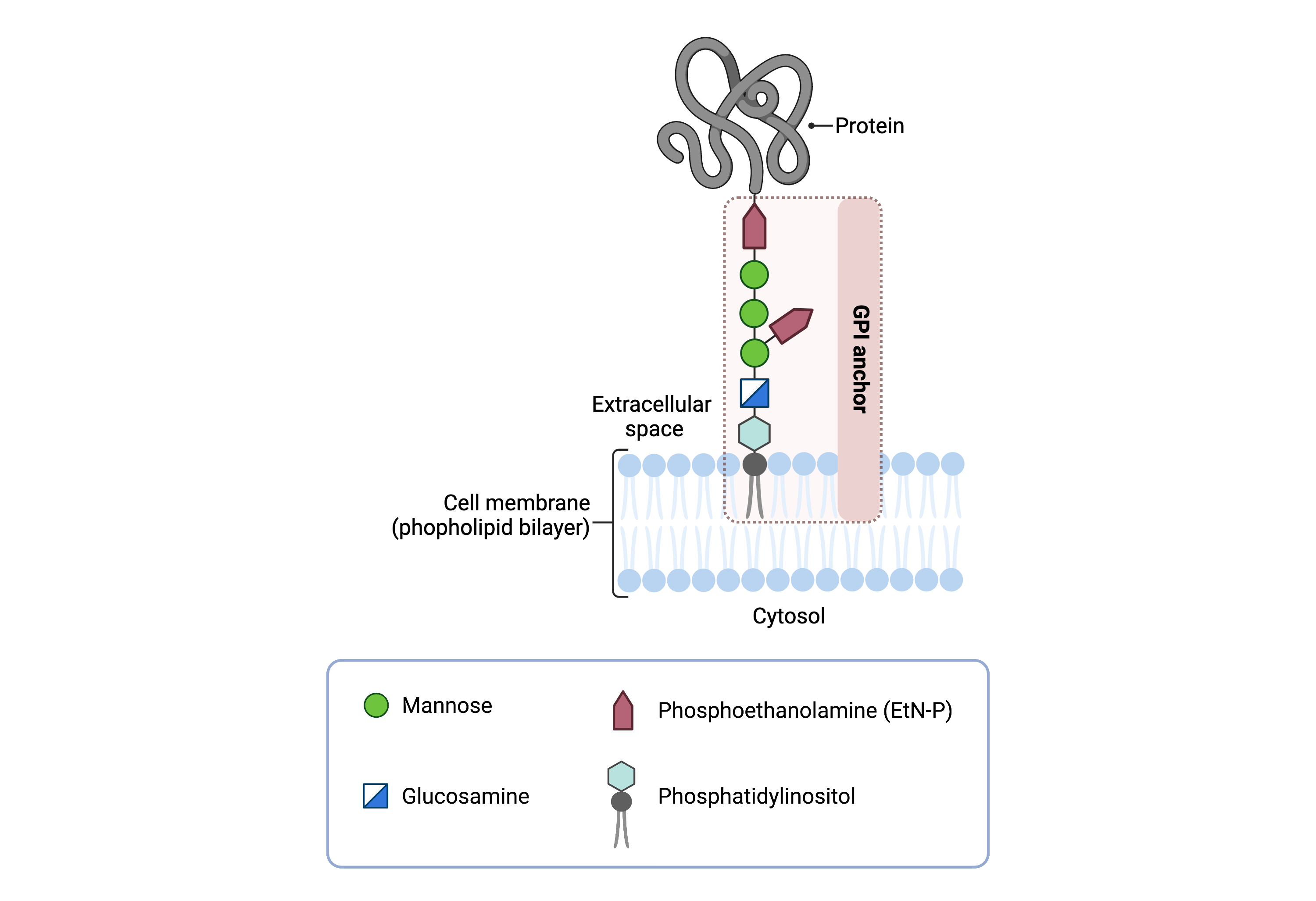
Figure 1: GPI-anchored protein structure.
The GPI anchor is a glycolipid that attaches proteins to the plasma membrane. The mammalian GPI core consists of an EtNP-Man-3-Man-2-Man-1-GlcN-PI backbone, with an EtN-P side branch linked to Man-1. The phosphatidylinositol lipid tail is embedded in the plasma membrane, where it associates with lipid rafts, facilitating protein localization and function.
The generation of a GPI-anchored protein is a multi-step process involving more than 30 enzymes and can be divided into the following steps (Figure 2)10–12:
- GPI anchor synthesis
- Protein attachment
- Lipid/glycan remodelling and protein transport
GPI anchor synthesis and protein attachment is largely carried out by a series of enzymes encoded by the PIG genes and the GPI-TA enzyme complex, while enzymes encoded by the PGAP genes facilitate remodelling of the GPI anchored protein.
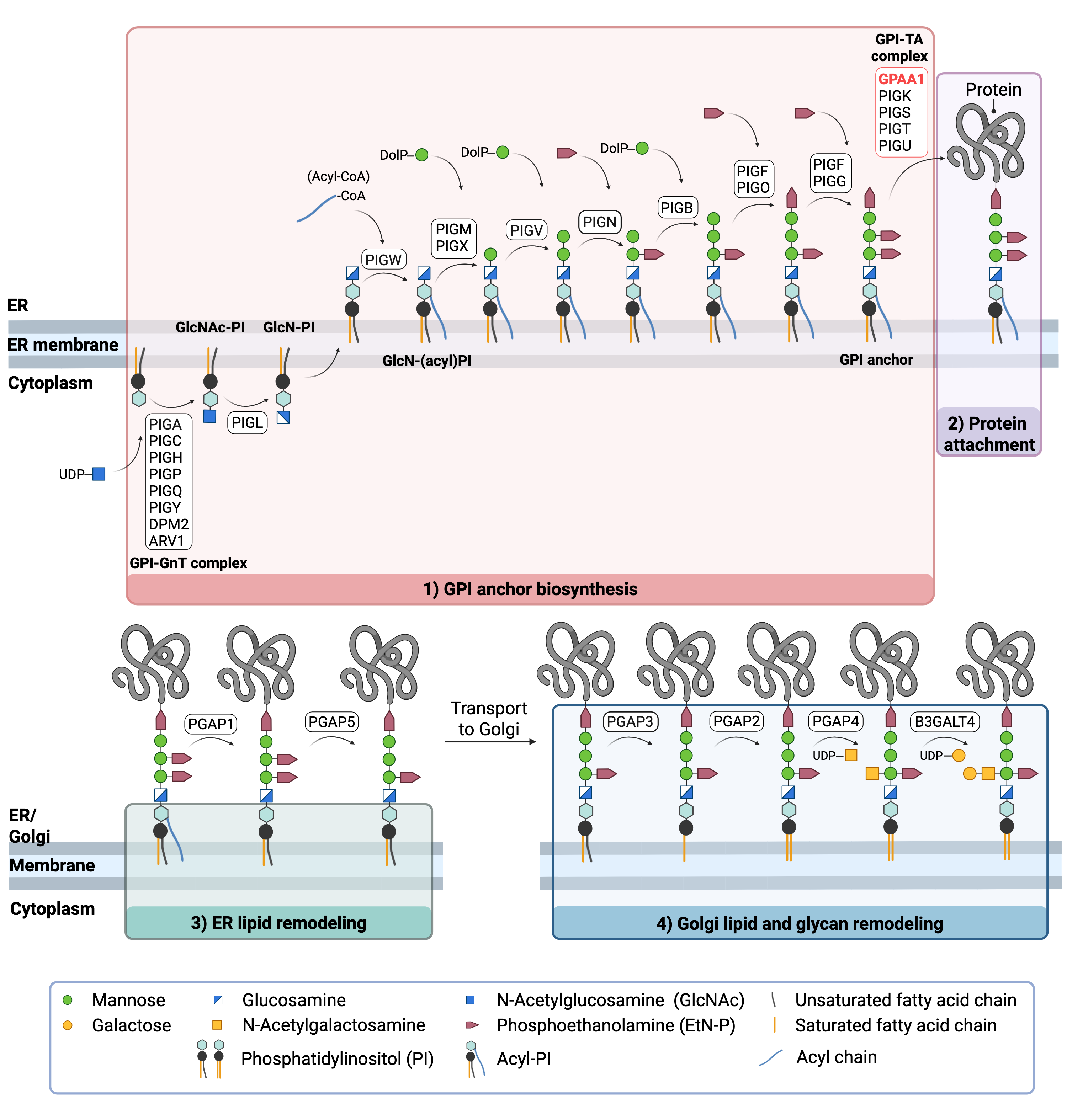
Figure 2: Overview of GPI-anchored protein biosynthesis and GPAA1 function.
GPI-anchored protein biosynthesis involves a series of enzymatic reactions. Beginning on the cytoplasmic side of the ER, the GPI anchor is synthesized in a stepwise process and then attached to target proteins. GPAA1 facilitates attachment of the GPI anchor to the protein by catalyzing the formation of a peptide bond between the EtN-P group of the GPI and the C-terminal of the protein. After protein attachment, the glycan and lipid components of the GPI anchor undergo further modifications in the ER and Golgi, before the mature GPI-AP is transported to the cell surface.
GPI ANCHOR SYNTHESIS
The first stage of GPI anchor biosynthesis involves the stepwise construction of the GPI anchor in the ER. This process begins on the cytoplasmic side of the ER, where N-acetyl glucosamine (GlcNAc) is transferred to the lipid phosphatidylinositol (PI) by the GPI GlcNAc transferase (GPI-GnT) complex, forming GlcNAc-PI8,13. The acetyl group from GlcNAc-PI is removed, generating glucosamine-phosphatidylinositol (GlcN-PI), which is then flipped from the cytoplasmic side to the luminal side of the ER membrane. Within the ER lumen, a fatty acid chain, commonly palmitic acid, is added by PIGW to form GlcN-(acyl)PI. Subsequent steps involve the sequential addition of three to four mannose sugars and three phosphoethanolamine (EtN-P) groups by several additional PIG enzymes11,14.
PROTEIN ATTACHMENT
Once the GPI anchor has been synthesized, it is transferred en bloc to a protein with a C-terminal GPI attachment signal sequence by GPI-TA. GPI-TA is a large enzyme complex comprised of 5 proteins: GPAA1, PIGK, PIGS, PIGT, and PIGU. Upon recognition of the signal sequence on the target protein, GPI-TA catalyzes the simultaneous cleavage of the signal sequence and attachment of the GPI anchor to the newly synthesized protein. GPAA1 carries out this protein attachment step by catalyzing the formation of a peptide bond between the phosphoethanolamine group of the GPI anchor and the C-terminal of the target protein11,15.
LIPID/GLYCAN REMODELLING AND PROTEIN TRANSPORT
After the protein has been attached to the GPI anchor, both the glycan and lipid portion of the anchor undergo modifications (referred to as remodelling) in the ER and Golgi by post-GPI attachment to protein (PGAP) enzymes or the glycosyltransferase B3GALT4. Following remodelling, the mature GPI-anchored protein is transported to the plasma membrane where it associates with other GPI-anchored proteins in lipid rafts16.
Disease Mechanism
Biallelic mutations in the GPAA1 gene results in partial loss of GPAA1 protein function. Some variants reduce GPAA1 mRNA expression, while others destabilize the protein1,2. Impaired GPAA1 function disrupts the final step of GPI anchor biosynthesis, preventing GPI anchor from being attached to target proteins. This leads to reduced expressed of GPI-APs on the cell surface1,2, and the accumulation of free GPI anchors in the ER15. GPI-APs are enriched at lipid raft domains in neurons, where they support overall neural development and function17. Their deficiency in GPAA1-CDG likely disrupts these processes, contributing to the disorder’s neurological features1.
Decreased levels of alkaline phosphatase (ALP), a GPI anchored enzyme that promotes bone mineralization by hydrolyzing phosphate compounds, may also contribute to reduced bone formation and density, resulting in osteopenia1,2. In GPI biosynthesis disorders that affect later steps of the GPI biosynthesis pathway, partially assembled GPI anchors can still trigger cleavage of the signal sequence on ALP by the GPI-TA complex, but the GPI anchor is not successfully attached. This results in secretion of unanchored ALP into the bloodstream, causing elevated serum ALP levels (hyperphosphatasia)18,19.
In contrast, mutations in subunits of the GPI-TA complex, such as GPAA1, prevent the attachment of fully assembled GPI anchors to target proteins, leading to retention and degradation of unanchored ALP within the ER, rather than its secretion18,19. As a result, individuals with CDG caused by variants in the GPI-TA complex typically exhibit normal or low serum ALP levels2. In GPAA1-CDG, it has been hypothesized that impaired anchoring of ALP may reduce its availability on the surface of osteoblasts, potentially disrupting bone mineralization and contributing to the development of osteopenia1,2. To date, no individuals with complete biallelic loss-of-function mutations in GPAA1 have been reported, and it has been proposed that residual GPAA1 activity may be sufficient to maintain near-normal serum ALP levels1. This is consistent with findings in most reported GPAA1-CDG cases, in which serum ALP has been described as normal1–3,7.
Mutations
The GPAA1 gene is located on Chromosome 8 (8q24.3). To date, more than 20 different variants have been reported in GPAA1-CDG patients, including missense, frameshift, splice-site, and nonsense. Several GPAA1 variants have been associated with specific effects on GPAA1 expression and protein stability. Some reported frameshift and splice site mutations appear to cause a decrease in GPAA1 mRNA expression based on analysis of patient samples. The p.Ala389Pro missense variant, located in the second transmembrane domain, has been found to result in GPAA1 protein instability in vitro1.
No homozygous loss-of-function variants have been identified thus far, suggesting the necessity of residual GPAA1 gene function2. The p.Trp76Ser and p.Leu291Pro missense variants have been found to sufficiently rescue GPI-AP activity in vitro, and the p.Trp176Ser variant has specifically been associated with a milder biochemical presentation as it does not show decreased activity1.
Signs & Symptoms
Clinical Presentation
Individuals with GPAA1-CDG typically develop signs and symptoms between early infancy and early to mid-childhood. GPAA1-CDG is primarily characterized by developmental delay, intellectual disability, cerebellar atrophy, low muscle tone (hypotonia), difficulties with muscle control, and abnormal eye movements. Reported symptoms of GPAA1-CDG include1–4,6:
- Neurological – global developmental delay, intellectual disability, low muscle tone (hypotonia), cerebellar atrophy, seizures, loss of muscle coordination (ataxia) including inability to perform controlled movements (dysmetria), weakness of muscles that control speech (dysarthria)
- Ophthalmological – nystagmus
- Skeletal – loss of bone density (osteopenia)
- Dysmorphic features – features vary widely across patients; prominent forehead, tented upper lip, and broad nose are common
Some affected individuals have presented with delayed protective coating of nerve cells (myelination), and mild thinning or incomplete development (hypoplasia) of the corpus callosum2,3.
Biochemical Presentation
GPAA1-CDG patients often display decreased GPI-AP expression levels at the cell surface based on flow cytometry analysis of skin and blood samples1,2. However, reports vary on the type of GPI-APs affected; some patient samples display reduced expression of CD16 and CD59 GPI-APs while others only show decreased expression of CD16.
In addition, most reported GPAA1-CDG patients have shown normal serum ALP levels1–3,7, except for one patient in which ALP levels were found to be low2.
Classification
GPAA1-CDG is classified as a disorder of GPI anchor biosynthesis.
Diagnosis
GPAA1-CDG should be suspected in individuals presenting with symptoms such as developmental delay, intellectual disability, hypotonia, seizures, dysmorphic features, and in many cases, cerebellar atrophy, osteopenia, and impaired motor coordination. All reported cases to date have been diagnosed through genetic testing, primarily exome sequencing1–6. Diagnosis may be supported by flow cytometry analysis of skin and blood cells obtained from patient samples to assess surface GPI-AP expression levels1,2. Additional evaluations often include brain MRI to assess structural abnormalities and EEG to detect seizure activity1–4.
Flow Cytometry
GPAA1-CDG patients have generally displayed reduced GPI-AP expression levels on the surface of blood and skin cells, including a decrease in CD16 and CD59 GPI-APs, as well as total GPI-AP expression levels by FLAER staining1. However, some patients have only presented with decreased CD16 expression levels, suggesting that the trend in GPI-AP deficiency is not yet completely clear and may vary based on the type of GPI-AP2.
MRI
Brain Magnetic Resonance Imaging (MRI) findings in individuals with GPAA1-CDG have often shown cerebellar atrophy1–3. Additional abnormalities may include delayed myelination and hypoplasia or thinning of the corpus callosum2.
EEG
Electroencephalography (EEG) findings in GPAA1-CDG patients have identified a range of seizure types, including febrile, generalized tonic-clonic, absence, myoclonic, and reflex seizures2,4.
Biomarkers
Flow cytometry analysis of skin and blood cells taken from patient samples (fibroblasts, leukocytes, and granulocytes) has largely shown decreased levels of GPI-AP expression, particularly CD16, CD73, and CD1091,2. Although surface expression of CD59 and total GPI-APs has been shown to vary between GPAA1-CDG patients, measuring surface GPI-AP expression is a suitable method of examining the functional impact of GPAA1 mutations2.
Serum ALP levels are often used as biomarkers for disorders affecting the GPI biosynthesis pathway. ALP is a GPI-AP that is normally found on the cell surface but may be secreted as a soluble protein or be insufficiently expressed on the cell surface depending on the GPI deficiency. Normal serum ALP levels have been reported in the majority of GPAA-CDG patients1–3,7, although they have appeared to be lower in one case2.
Prognosis
Prognosis of GPAA1-CDG may vary depending on the severity of an individual’s symptoms. Due to the rarity of GPAA1-CDG, its long-term prognosis and the life expectancy of affected individuals is currently unknown. Two patients have shown progressive shrinking of the cerebellum from infancy to early childhood, and seizures typically begin between 8 months and 3 years2,3. As of 2021, the oldest reported patient is 38 years of age2.
Management
Symptoms may be managed through a combination of physical, occupational, and speech therapy, especially in addressing motor developmental delay and speech difficulties. Various drug and diet-based interventions have been reported to control seizures in GPAA1-CDG patients, including antiepileptic medications as well as a ketogenic diet, which was found to be partially effective in some individuals2,3. Pyridoxine supplementation may help manage seizures among individuals affected by GPI biosynthesis defects, however further testing is required to determine the effectiveness of this treatment for GPAA1-CDG1.
Therapies
There are currently no direct treatment options available for GPAA1-CDG.
Research Models
There are several available research models of GPAA1-CDG, including yeast, fly, zebrafish, and mouse models, as well as human cell lines.
Yeast (S. cerevisiae)
GAA1 is the ortholog of the mammalian GPAA1 gene in the S. cerevisiae yeast species (SGD). It is an essential gene in yeast as its deletion results in lethality20. A temperature-sensitive GAA1 mutant yeast strain (TSA207), which is lethal at 38.5°C, is available (Euroscarf).
The mammalian and yeast orthologs share 25% amino acid identity21. Similar to the mammalian GPAA1 protein, yeast Gaa1p is responsible for attaching proteins to GPI anchors in the ER, and mutant cells show a lack of GPI-AP expression at the cell surface20.
Worm (C. elegans)
gpaa-1 is the C. elegans ortholog of the mammalian GPAA1 gene (WormBase). The GPAA-1 protein is predicted to be part of the GPI-TA complex and involved in the attachment of the GPI anchor to a protein.
Fly (D. melanogaster)
gaa1 is the Drosophila ortholog of the mammalian GPAA1 gene (FlyBase). It encodes a protein that is part of the GPI-TA complex which is active in the ER and nuclear membrane. It is located in the endomembrane system and is involved in GPI anchor biosynthesis and biogenesis of the rhabdomere membrane (located in the eye). It is expressed in the adult head, embryonic and larval midgut, as well as the ganglia (Alliance Genome). It is an essential gene in flies; null mutants die before the end of the larval stage22.
Zebrafish (D. rerio)
gpaa1 is the zebrafish ortholog of the mammalian GPAA1 gene (ZFIN). Gpaa1 is predicted to be part of the GPI-TA complex and be involved in the attachment of the GPI anchor to a protein. A zebrafish gpaa1 knockout model was generated through morpholino microinjection and resulted in developmental defects including shortened body, back hyperextension, underdeveloped eyes, float deficiency, fluid buildup in the space around the heart (pericardial edema), and thickening of artery walls (vascular dysplasia)23.
Mouse (M. musculus)
Gpaa1-/- knockout mice
Homozygous Gpaa1-knockout mice carrying biallelic exon deletion mutations were generated through the CRISPR-Cas9 system. Significant phenotypes included embryonic lethality before the tooth bud stage and preweaning lethality (no survival past day 21 of the postnatal stage) (IMPC).
Gpaa1+/- knockout mice
Heterozygous Gpaa1-knockout mice carrying an exon deletion mutation were generated through the CRISPR-Cas9 system. Significant phenotypes included abnormal embryo size, abnormal retina vasculature morphology, and embryonic growth retardation (IMPC).
Gpaa1-/- embryonal carcinoma cell line
Homozygous Gpaa1-knockout mouse embryonal carcinoma cells were generated in vitro. These knockout cells displayed a lack of surface GPI-AP expression and accumulation of mature GPI anchors in the ER, indicating a block in GPI anchor attachment to proteins24.
Human Cell Lines
GPAA1-/- HEK293 knockout cells
HEK293 GPAA1-knockout cells were generated using the CRISPR-Cas9 system to study the role of GPAA1 in the GPI-TA complex. GPAA1-knockout cells exhibit a reduced expression of CD55. To investigate functionally important residues, a panel of GPAA1 mutants was expressed in the knockout cells. Two knockout variants, p.Ser51Leu and p.Ala389Pro, showed reduced GPI-TA activity compared to wild-type GPAA1. The p.Ala389Pro variant was specifically reported to result in protein instability. p.Trp176Ser and p.Leu291Pro were predicted to retain partial activity. Asp250 on GPAA1 was identified as a critical catalytic residue required for GPI-TA enzymatic activity1.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a natural history study on all CDG types, including GPAA1-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life
Organizations & Groups
GPI-anchor CDG Community Facebook Group
Publications
GPAA1-CDG Scientific Articles on PubMed
Additional Resources
OMIM
IEMbase
OrphaNet
NORD
Genetic Testing Registry
ClinVar
NIH
GeneCards
UniProt
References
- Nguyen, T. T. M. et al. Mutations in GPAA1, Encoding a GPI Transamidase Complex Protein, Cause Developmental Delay, Epilepsy, Cerebellar Atrophy, and Osteopenia. The American Journal of Human Genetics 101, 856–865 (2017).
- Castle, A. M. R. et al. Expanding the Phenotypic Spectrum of GPI Anchoring Deficiency Due to Biallelic Variants in GPAA1. Neurol Genet 7, (2021).
- Fontana, P. et al. A Novel Homozygous GPAA1 Variant in a Patient with a Glycosylphosphatidylinositol Biosynthesis Defect. Genes (Basel) 14, (2023).
- Chen, Q. R., Zhang, Z. J., Lu, Y. X., Yuan, S. B. X. & Li, J. Glycosylphosphatidylinositol biosynthesis deficiency 15 caused by GPAA1 gene mutation: a rare disease study. Chinese Journal of Contemporary Pediatrics 25, 1276–1281 (2023).
- Mahdiannasser, M., Rashidi-Nezhad, A., Badv, R. S. & Akrami, S. M. Exploring the genetic etiology of drug-resistant epilepsy: incorporation of exome sequencing into practice. Acta Neurol Belg 122, 1457–1468 (2022).
- Bibi, A. et al. Exome sequencing reveals genetic heterogeneity in consanguineous Pakistani families with neurodevelopmental and neuromuscular disorders. Am J Med Genet C Semin Med Genet 196, e32103 (2024).
- Sidpra, J. et al. The clinical and genetic spectrum of inherited glycosylphosphatidylinositol deficiency disorders. Brain 147, 2775–2790 (2024).
- Kinoshita, T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biol 10, 190290 (2020).
- Knaus, A. et al. Mutations in PIGU Impair the Function of the GPI Transamidase Complex, Causing Severe Intellectual Disability, Epilepsy, and Brain Anomalies. Am J Hum Genet 105, 395–402 (2019).
- Kinoshita, T. & Fujita, M. Thematic Review Series: Glycosylphosphatidylinositol (GPI) Anchors: Biochemistry and Cell Biology Biosynthesis of GPI-anchored proteins: special emphasis on GPI lipid remodeling. J Lipid Res 57, 6–24 (2016).
- Liu, Y.-S. & Fujita, M. Mammalian GPI-anchor modifications and the enzymes involved. Biochem Soc Trans 48, 1129–1138 (2020).
- Englund, P. T. The Structure and Biosynthesis of Glycosylphosphatidylinositol Protein Anchors. Annu Rev Biochem 62, 121–138 (1993).
- Watanabe, R. et al. The first step of glycosylphosphatidylinositol biosynthesis is mediated by a complex of PIG‐A, PIG‐H, PIG‐C and GPI1. EMBO J 17, 877-885–885 (1998).
- Maeda, Y. & Kinoshita, T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Prog Lipid Res 50, 411–424 (2011).
- Eisenhaber, B., Eisenhaber, S., Kwang, T. Y., Grub̈er, G. & Eisenhaber, F. Transamidase subunit GAA1/GPAA1 is a M28 family metallo-peptide-synthetase that catalyzes the peptide bond formation between the substrate protein’s omega-site and the GPI lipid anchor’s phosphoethanolamine. Cell Cycle 13, 1912–1917 (2014).
- Fujita, M. & Kinoshita, T. Structural remodeling of GPI anchors during biosynthesis and after attachment to proteins. FEBS Press 584, 1670–1677 (2009).
- Um, J. W. & Ko, J. Neural Glycosylphosphatidylinositol-Anchored Proteins in Synaptic Specification. Trends Cell Biol 27, 931–945 (2017).
- Murakami, Y. et al. Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome. Journal of Biological Chemistry 287, 6318–6325 (2012).
- Thompson, M. D. & Knaus, A. Rare Genetic Developmental Disabilities: Mabry Syndrome (MIM 239300) Index Cases and Glycophosphatidylinositol (GPI) Disorders. Genes (Basel) 15, (2024).
- Hamburger, D., Egerton, M. & Riezman, H. Yeast Gaa1p is required for attachment of a completed GPI anchor onto proteins. Journal of Cell Biology 129, 629–639 (1995).
- Hiroi, Y. et al. Molecular cloning of human homolog of yeast GAA1 which is required for attachment of glycosylphosphatidylinositols to proteins1The sequence reported in this paper has been doposited in the GenBank, EMBL and GenBank nucleotide sequence databases with the accession number AB006969.1. FEBS Lett 421, 252–258 (1998).
- Satoh, T., Inagaki, T., Liu, Z., Watanabe, R. & Satoh, A. K. GPI biosynthesis is essential for rhodopsin sorting at the trans-Golgi network in Drosophila photoreceptors. Development (Cambridge) 140, 385–394 (2013).
- Li, Y. et al. A novel variant in GPAA1, encoding a GPI transamidase complex protein, causes inherited vascular anomalies with various phenotypes. Hum Genet 139, 1499–1511 (2020).
- Ohishi, K. et al. Gaa1p and Gpi8p Are Components of a Glycosylphosphatidylinositol (GPI) Transamidase That Mediates Attachment of GPI to Proteins. Mol Biol Cell 11, 1523–1533 (2000).