
Lay Summary
DPAGT1-CDG, formerly known as CDG-Ij, is a rare inherited condition that affects many systems in the body. To date, 40 patients are reported in the medical literature. DPAGT1-CDG is classified as a disorder of N-linked protein glycosylation. DPAGT1-CDG is caused when an individual has mutations in both copies of their DPAGT1 gene, which provides instructions for making an enzyme that catalyzes the first step in the N-glycosylation pathway. Mutations in the DPAGT1 result in impaired formation of N-linked glycoproteins. DPAGT1-CDG patients may have a severe presentation, characterized by neurological abnormalities, intellectual disability, epilepsy, small head size, and low muscle tone, or a mild presentation primarily consisting of a neuromuscular transmission disorder associated with muscle weakness and fatigue; the mild presentation is also known as DPAGT1 congenital myasthenic syndrome (DPAGT1-CMS). Screening tests are available for DPAGT1-CDG, but a definitive diagnosis is achieved through genetic testing. There are currently no approved treatments for DPAGT1-CDG, but acetylcholinesterase inhibitors have been used to treat muscle-related symptoms in patients with both mild and severe disease. Other treatments are focused on the management of specific symptoms and preventing complications.
Overview
Dolichyl-phosphate N-Acetylglucosamine (GlcNAc) phosphotransferase 1 congenital disorder of glycosylation (DPAGT1-CDG) is a rare autosomal recessive genetic disorder. The DPAGT1 gene encodes an enzyme that is responsible for attaching GlcNAc-phosphate to the lipid carrier dolichol; this is the first step in the synthesis of the lipid-linked oligosaccharide (LLO) in the endoplasmic reticulum (ER) (Figure 1)1,2. LLO synthesis is a precursor step to N-glycosylation, which is the process by which sugar chains (glycans) are added to the amino acid asparagine in some proteins3. Many proteins require N-glycosylation to achieve full functionality. Mutations to DPAGT1 result in an enzyme with reduced catalytic activity, where GlcNAc transfer to dolichol is reduced or absent. Deficiency in the DPAGT1 enzyme results in the incomplete assembly of the LLO, leading to insufficient N-glycosylation of glycoproteins1.
The first reported case of DPAGT1-CDG was in 2003 and there are 40 confirmed cases to date, 18 of which are members of a large consanguineous family2,4–7. Symptoms typically begin at infancy, but some cases show symptoms during pregnancy with decreased fetal activity. Symptom onset may also be delayed into childhood. Characteristic presentations of DPAGT1-CDG with onset at infancy include severe neurological symptoms, gastrointestinal and feeding problems, and dysmorphic features2,4–7.
Several genes associated with CDG (DPAGT1, ALG2, ALG14, GFPT1) are also associated with congenital myasthenic syndrome (CMS), a group of neuromuscular transmission disorders8,9. DPAGT1-associated congenital myasthenia syndrome (DPAGT1-CMS) is a less severe presentation of DPAGT1-CDG and is characterized by muscle weakness and fatigue beginning in childhood10. A potential diagnosis of DPAGT1-CDG can be determined through transferrin analysis, but a definitive diagnosis can only be achieved through genetic testing. There are currently no approved treatments for DPAGT1-CDG.
Synonyms
- CDG-Ij; CDG IIJ
- CDG-1J; CDG 1J
- CDG syndrome type Ij
- Congenital disorder of glycosylation type Ij
- Carbohydrate deficient glycoprotein syndrome type Ij
- DPAGT1-associated congenital myasthenia syndrome; DPAGT1-CMS
- ALG7-CDG
Inheritance
DPAGT1-CDG is an autosomal recessive disorder, meaning an affected individual inherits one defective copy of the gene from each asymptomatic parent.
Gene Function
The DPAGT1 gene encodes the enzyme UDP-GlcNAc: dolichylphosphate N-Acetylglucosaminephosphotransferase 1 (DPAGT1). The DPAGT1 enzyme is also called GlcNAc-1-P Transferase or ALG7. DPAGT1 is located in the ER membrane where it has a role in the assembly of the LLO, the oligosaccharide precursor used in protein N-glycosylation1. The DPAGT1 enzyme catalyzes the first step in LLO synthesis: the transfer of GlcNAc-phosphate from the nucleotide sugar, UDP-GlcNAc, to the lipid carrier dolichol-phosphate (Dol-P). Successful LLO synthesis is a prerequisite for N-glycosylation, wherein the glycan chain is transferred from the LLO to an asparagine residue on a protein.
LLO synthesis
N-glycosylation is the process by which an oligosaccharide is attached to the nitrogen atom of asparagine residues on proteins. N-glycosylation is initiated in the ER and begins with the synthesis of the LLO3,11. The LLO is comprised of a 14-sugar oligosaccharide, which is sometimes referred to as the N-glycan precursor oligosaccharide, attached to the lipid carrier dolichol pyrophosphate (Dol-PP). The 14-sugar glycan chain is made up of 2 N-acetylglucosamine, 9 mannose, and 3 glucose residues. Once assembled, the oligosaccharide is transferred en bloc to proteins and undergoes further processing in the ER and Golgi3,11. Once the oligosaccharide is attached to a protein, it is referred to as an N-glycan.
LLO synthesis is carried out by a series of enzymes encoded by the DPAGT1 and ALG genes and can be divided into two phases: Phase I and Phase II (Figure 1)3.
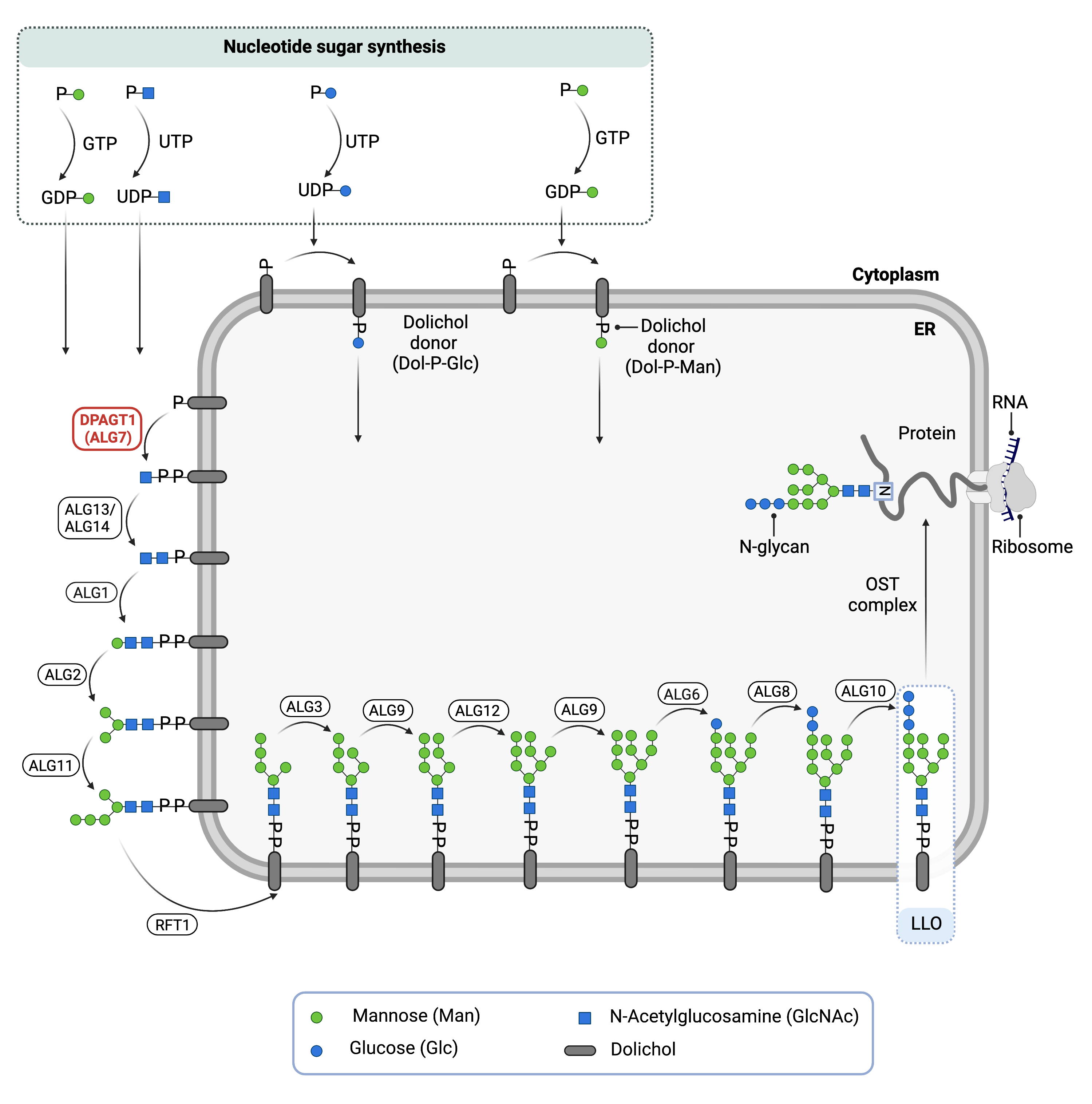
Figure 1. The Role of DPAGT1 in glycosylation.
DPAGT1 is an enzyme (GlcNAc-1-P Transferase) that is involved in synthesizing the lipid-linked oligosaccharide (LLO) in N-glycosylation. DPAGT1 catalyzes the transfer of GlcNAc-phosphate from the nucleotide sugar, UDP-GlcNAc, to Dol-P, generating Dol-PP-GlcNAc.
Phase I
Phase I of LLO synthesis takes place on the cytoplasmic side of the ER. It begins with the sequential attachment of two N-Acetylglucosamine (GlcNAc2) and five mannose (Man5) residues to Dol-PP by several ER enzymes. The DPAGT1 enzyme catalyzes the transfer of GlcNac-phosphate from the nucleotide sugar, UDP-GlcNAc, to Dol-P, generating Dol-PP-GlcNAc. The subsequent GlcNAc and mannose sugars are transferred from UDP-GlcNAc and GDP-mannose onto the growing LLO by several enzymes. The intermediate structure, Dol-PP-GlcNAc2Man5, is translocated from the cytoplasm into the ER lumen by the RFT1 enyzme3,11 .
Phase II
Phase II of LLO synthesis takes place in the ER lumen. Once in the lumen, four mannose residues followed by three glucose (Glc3) residues are added to the intermediate structure, generating the complete LLO, Dol-PP-GlcNAc2Man9Glc3. Mannose and glucose are transferred from glycosyl donors Dol-P-mannose and Dol-P-glucose, which are formed on the cytoplasmic side of the ER and must also be flipped across the ER membrane11.
Once assembled, the oligosaccharide is transferred en bloc from Dol-PP to asparagine residues of newly synthesized protein via the enzyme complex oligosaccharyltransferase (OST), resulting in N-glycosylation of the protein13. The activity of OST is highly specific for the completely assembled 14-sugar oligosaccharide, GlcNAc2Man9Glc3.
Disease Mechanism
Mutations in the DPAGT1 gene lead to the production of an abnormal enzyme with reduced or no activity, thereby disrupting the transfer of the first GlcNAc residue onto Dol-P1. As a result, LLO synthesis is incomplete and the transfer of its glycan component onto proteins is impaired, resulting in N-glycoproteins that are insufficiently glycosylated (hypoglycosylation). Since N-glycans are important for the proper function of many processes in the body, DPAGT1-CDG affects multiple systems. In DPAGT1-CMS, the DPAGT1 enzyme may retain a larger percentage of normal enzymatic activity which mitigates the multisystemic phenotype1. It is proposed that the primary disease mechanism in DPAGT1-CMS is reduced levels of acetylcholine receptors at the endplate region of the neuromuscular junction as a result of hypoglycosylation10.
Mutations
The DPAGT1 gene is located on Chromosome 11 (11p23.3) and 25 disease causing mutations have been reported for the DPAGT1 gene including 21 missense mutations, 3 splicing mutations, and one duplication mutation14.
Mutations in DPAGT1 causing CMS have been found in the conserved region of the protein, indication a disruption to proper functioning of the protein10. Patients with DPAGT1-CMS typically have compound heterozygous mutations that results in 50% or more normal catalytic activity on one allele, while individuals with DPAGT1-CDG have significant loss of function in both alleles. In both cases, mutations may either reduce the stability of the protein or reduce enzyme activity1.
Signs & Symptoms
Clinical Presentation
Individuals with DPAGT1-CDG typically have either a severe, multisystemic presentation or a milder presentation that primarily involves muscle weakness (DPAGT1-CMS).
DPAGT1-CDG
Individuals with DPAGT1-CDG typically develop signs and symptoms during pregnancy (decreased fetal activity) or infancy13. DPAGT1-CDG is primarily characterized by a multi-systemic disorder with mild to severe neurological impairment. The characteristic clinical presentations of DPAGT1-CDG include2,4–7:
- Neurological – global developmental delay, frequent seizures that get worse over time, cognitive impairment ranging from mild to severe, muscle weakness and low muscle tone (hypotonia), proximal muscle weakness, lack of coordination and muscle control (ataxia), and early-onset epileptic encephalopathy.
- Gastrointestinal – feeding difficulties and intestinal problems may lead to a failure to gain weight and slower than normal growth (failure to thrive). Protein-losing enteropathy (PLE) is one of the more life-threatening symptoms, causing an excess loss of proteins in the gastrointestinal tract.
- Dysmorphic features – misaligned eyes with an inward turning of one or both eyes (esotropia), small head (microcephaly), small lower jaw, narrow mouth, stiff joints (contractures), and bent pinky fingers.
- Brain structure abnormalities – underdeveloped cerebellum (cerebellar hypoplasia).
Other less common clinical presentations include speech problems, and behavioural problems.
DPAGT1-CMS
Individuals with DPAGT1-CMS typically present with symptoms in childhood which is primarily characterized by muscle weakness. The characteristic clinical presentations of DPAGT1-CMS include8,10:
- Neurological – severely affected proximal and distal limb muscle weakness, mildly affected facial, ocular, bulbar, and respiratory muscles
Patients with DPAGT1-CMS do not display the other multi-system symptoms associated with DPAGT1-CDG10.
Biochemical Abnormalities
Biochemical abnormalities observed in individuals with DPAGT1-CDG include blood clotting (coagulation) abnormalities, elevated levels of the muscle enzyme creatine kinase, and elevated liver enzymes (transaminases)14.
Classification
DPAGT1-CDG is classified as a disorder of N-linked protein glycosylation. Under the former CDG classification system DPAGT1-CDG is classified as a Type 1 CDG, which arise due to defects in the production of lipid-linked oligosaccharides or their transfer onto proteins during N-glycosylation.
Diagnosis
Although diagnosis of DPAGT1-CDG may be suspected based on presentation of symptoms and a detailed patient history, direct molecular genetic testing is the only definitive diagnostic test. Preimplantation screening has also been used to identify DPAGT1 mutations in at-risk embryos15. Screening in suspected patients begins with a blood test to analyze serum transferrin. Specialized testing on muscle and nerves is typically carried out when DPAGT1-CMS is suspected.
Transferrin Analysis
Individuals with DPAGT1-CDG show a type 1 pattern by transferrin isoelectric focusing (TIEF) or mass spectrometry analysis of transferrin6,7. Type 1 patterns are observed in CDG that arise due to defects in LLO assembly and are characterized by a decrease in tetrasialo-transferrin and an increase in di-sialo and a-sialo transferrin isoforms.
In patients with DPAGT1-CMS, transferrin patterns are reported as normal in approximately half of the patients tested16.
Muscle Biopsy
Tubular aggregates are observed in muscle biopsies of patients with DPAGT1-CMS10.
Single Fiber Electromyography (EMG)
For patients with DPAGT1-CMS, single fiber EMG or nerve stimulation tests can be used to investigate the activity of individual muscle fibers and detect abnormal signalling between nerves and muscles10.
Prognosis
Prognosis of DPAGT1-CDG may vary depending on the severity of an individual’s symptoms. Most patients with DPAGT1-CDG have severe symptoms and 80% die in early childhood from complications of early-onset epileptic encephalopathy and severely delayed development5–7. The oldest individual with DPAGT1-CDG that has been described in the medical literature was 34 years old, as reported in 20195.
Management
Management of DPAGT1-DG requires a multidisciplinary team and may include combinations of physical therapy, occupational therapy, speech or vision therapy and palliative measures14.
Therapies
It has been reported that acetylcholinesterase inhibitors may improve muscle related symptoms for patients with CDG and CMS10,14. Treatment for other symptoms is focused on management of symptoms and prevention of complications.
Research Models
Several DPAGT1 research models have been generated including yeast, fly, worm, mouse model organisms and patient-derived fibroblasts.
Yeast (S. cerevisiae)
Yeast ALG7 is orthologous to human DPAGT1. Null mutations to the ALG7 gene in yeast show that ALG7 is required for cell growth. Mutations that disrupt the ALG7 gene result in cell lethality17. Additionally, it was found that mutants have a difference in transcript number compared to wild-type, which may influence the synthesis of regulatory elements directly or indirectly.
Fly (D. melanogaster)
Drosophila gene Alg7 (Alg7, CG5287, FBgn0032477) is orthologous to DPAGT1, also encoding the UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosamine phosphotransferase enzyme. It is a potential model for the human disease DPAGT1-CDG (FlyBase).
Worm (C. elegans)
Algn-7 (orthologous to DPAGT1 in humans) was mutated using RNAi and deletion mutagenesis. It was found that Algn-7 and N-glycosylation are required for oogenesis and oocyte-to-embryo transition18. Mutant phenotypes observed included larval lethality, defects in oogenesis and oocyte-to-embryo transition. Five germline-expressed genes with similar phenotypes to algn-7 were identified by examining RNAi phenotypes, suggesting a tool to detect new CDG candidate genes and associated gene networks.
Mouse (M. musculus)
Homozygous Dpagt1-/- knockout mouse
Homozygous deletion of Dpagt1 in mouse embryos is embryonic lethal19.
Homozygous Dpagt1D166G/D166G missense mutation
A homozygous missense mutation (D166G) in Dpagt1 has been found to impede follicle development in female mice, resulting in less eggs at ovulation, as well as underdeveloped oocytes after in vitro fertilization20. Conditional knockouts of Dpagt1 in oocytes specifically have been shown to cause infertility.
Dpagt1 knockout ES cell line
Embryonic Stem (ES) cell line Dpagt1tm1a(EUCOMM)Hmgu is a targeted knockout/null mutation of Alg6. A “conditional-ready” allele can be created by flp recombinase expression in mice carrying this allele, and cre expression results in a knockout mouse. If cre is expressed without flp expression, a reporter knockout mouse can be generated (MGI).
Human Cell Lines
Patient-derived fibroblasts
Patient-derived fibroblasts have been used to examine DPAGT1 transcriptional profiles and nucleotide GTP levels2. A study showed that the six mutations analyzed (p.Leu120Met, p.Val264Gly, p.Arg301Cys, p.Arg301His, p.Leu385Arg, and p.Phe110Ser) affect either splicing activity, GTP stability, or the ability of the protein to localize to the ER membrane, as well as a reduced response to ER stress.
Clinical Studies
Active
Clinical and Basic Investigations into Congenital Disorders of Glycosylation (NCT04199000)
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including DPAGT1-CDG. The purpose of this study is to define the natural history and clinical symptoms of CDG, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Publications
DPAGT1-CDG Scientific Articles on PubMed
Additional Resources
DPAGT1-CDG Clinical Utility Gene Card
References
- Dong, Y. Y. et al. Structures of DPAGT1 Explain Glycosylation Disease Mechanisms and Advance TB Antibiotic Design. Cell 175, 1045-1058.e16 (2018).
- Yuste-Checa, P. et al. DPAGT1-CDG: Functional analysis of disease-causing pathogenic mutations and role of endoplasmic reticulum stress. PLoS One 12, e0179456 (2017).
- Stanley, P., Taniguchi, N. & Aebi, M. N-Glycans. in Essentials of Glycobiology (eds. Varki, A., Cummings, R. & Esko, J. et al.) (Cold Spring Harbor Laboratory Press, 2017). doi:10.1101/GLYCOBIOLOGY.3E.009.
- Wu, X. et al. Deficiency of UDP-GlcNAc:Dolichol Phosphate N-Acetylglucosamine-1 Phosphate Transferase (DPAGT1) Causes a Novel Congenital Disorder of Glycosylation Type Ij. Hum. Mutat. 22, 144–150 (2003).
- Ng, B. G. et al. DPAGT1 deficiency with encephalopathy (DPAGT1-CDG): Clinical and genetic description of 11 new patients. JIMD Rep. 44, 85–92 (2019).
- Würde, A. E. et al. Congenital disorder of glycosylation type Ij (CDG-Ij, DPAGT1-CDG): Extending the clinical and molecular spectrum of a rare disease. Mol. Genet. Metab. 105, 634–641 (2012).
- Imtiaz, F., Al-Mostafa, A. & Al-Hassnan, Z. N. Further delineation of the phenotype of congenital disorder of glycosylation DPAGT1-CDG (CDG-Ij) identified by homozygosity mapping. JIMD Rep. 2, 107–111 (2012).
- Cossins, J. et al. Congenital myasthenic syndromes due to mutations in ALG2 and ALG14. Brain 136, 944–956 (2013).
- Paprocka, J., Jezela-Stanek, A., Tylki-Szymańska, A. & Grunewald, S. Congenital Disorders of Glycosylation from a Neurological Perspective. Brain Sci. 2021, Vol. 11, Page 88 11, 88 (2021).
- Belaya, K. et al. Identification of DPAGT1 as a new gene in which mutations cause a congenital myasthenic syndrome. Ann. N. Y. Acad. Sci. 1275, 29–35 (2012).
- Bieberich, E. Synthesis, Processing, and Function of N-glycans in N-glycoproteins. in Glycobiology of the Nervous System vol. 9 47–70 (Springer, 2014).
- Harada, Y., Ohkawa, Y., Kizuka, Y. & Taniguchi, N. Oligosaccharyltransferase: A Gatekeeper of Health and Tumor Progression. Int. J. Mol. Sci. 2019, Vol. 20, Page 6074 20, 6074 (2019).
- Carrera, I. A., Matthijs, G., Perez, B. & Cerdá, C. P. DPAGT1-CDG: Report of a patient with fetal hypokinesia phenotype. Am. J. Med. Genet. Part A 158A, 2027–2030 (2012).
- Jaeken, J., Lefeber, D. & Matthijs, G. Clinical utility gene card for: DPAGT1 defective congenital disorder of glycosylation. Eur. J. Hum. Genet. 23 (2015) doi:10.1038/ejhg.2015.177.
- Tabatabaei, Z. et al. Pre-Implantation Genetic Testing for Monogenic Disorders(PGT-M) in A Family with A Novel Mutationin DPAGT1 Gene. Cell J. 23, 593 (2021).
- DPAGT1-congenital disorder of glycosylation (DPAGT1-CDG). Frontiers in Congenital Disorders of Glycosylation https://www.rarediseasesnetwork.org/fcdgc/dpagt1.
- Kukuruzinska, M. A. & Robbins, P. W. Protein glycosylation in yeast: Transcript heterogeneity of the ALG7 gene. Proc. Natl. Acad. Sci. U. S. A. 84, 2145–2149 (1987).
- Kanaki, N. et al. UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase is indispensable for oogenesis, oocyte-to-embryo transition, and larval development of the nematode Caenorhabditis elegans. Glycobiology 29, 163–178 (2019).
- Marek, K. W., Vijay, I. K. & Marth, J. D. A recessive deletion in the GlcNAc-1-phosphotransferase gene results in peri-implantation embryonic lethality. Glycobiology 9, 1263–1271 (1999).
- Li, H. et al. DPAGT1-Mediated Protein N-Glycosylation Is Indispensable for Oocyte and Follicle Development in Mice. Adv. Sci. 7, 2000531 (2020).