
Lay Summary
DOLK-CDG, formerly known as CDG-Im, is a rare inherited condition that affects several body systems. To date, approximately 20 cases of DOLK-CDG have been reported in the literature. DOLK-CDG is classified as a disorder of multiple glycosylation pathways and can be subcategorized as a disorder of dolichol metabolism. It is caused when an individual has mutations in both copies of the DOLK gene, which provides instructions for making an enzyme that transfers a phosphate molecule onto the newly synthesized dolichol – a lipid that is needed for different types of glycosylation. Mutations in the DOLK gene reduce the availability of dolichol, leading to proteins with abnormal sugar chains. Symptoms of DOLK-CDG are highly variable and can range from only cardiac involvement (weakened and enlarged heart), only neurological involvement and vision problems (intellectual disability, autism spectrum disorder, and seizures), or in severe cases, affecting multiple body systems. In each clinical presentation, skin symptoms are also common. A screening test is available for DOLK-CDG but a definitive diagnosis is achieved through genetic testing. Some patients with mild symptoms have successfully received heart transplants, which have the potential to be curative. Other treatment focuses of managing specific symptoms and preventing complications.
Overview
Dolichol kinase congenital disorder of glycosylation (DOLK-CDG) is a rare autosomal recessive genetic disorder. The first confirmed case of DOLK-CDG was reported in 20071 and there are approximately 20 cases reported in the literature to date1–7.
The DOLK gene encodes dolichol phosphate (DOLK), an enzyme that catalyzes the transfer of a phosphate molecule onto dolichol following dolichol biosynthesis. Biosynthesis of dolichol is one of the first steps in glycosylation, where dolichol acts as a lipid carrier for the oligosaccharide precursor in the early stages of N-glycosylation of proteins. Dolichol is also a carrier for monosaccharides (mannose and glucose) used as donors in O-mannosylation, C-mannosylation and GPI anchor biosynthesis. Mutations in the DOLK gene result in hypoglycosylation of N-glycoproteins and may cause defects in multiple glycosylation pathways1,2,8,9
Symptoms of DOLK-CDG begin in infancy and can be highly variable, falling into three clinical presentations; (1) cardiac symptoms including a weakened and enlarged heart, (2) neurological symptoms including recurrent seizures, and intellectual disability, and (3) severe multisystem involvement. All three clinical presentations can also include dry and scaly skin (ichthyosis)3. Symptoms vary depending on type of mutation and the severity of the DOLK deficiency1–7. A definitive diagnosis can only be achieved through genetic testing, but transferrin analysis typically shows a type I pattern. Patients with mild symptoms of DOLK-CDG have received successful heart transplants that may be curative10,11.
Synonyms
- CDG-Im
- Congenital disorder of glycosylation, type Im
- Dolichol kinase deficiency
- DK1 deficiency
Inheritance
DOLK-CDG is inherited in an autosomal recessive fashion, with one mutation inherited from each asymptomatic (carrier) parent.
Gene Function
The DOLK gene encodes the enzyme dolichol kinase (DOLK), which is also called DK1. DOLK catalyzes the transfer of phosphate from cytidine triphosphate (CTP) to dolichol following biosynthesis of dolichol, generating dolichol phosphate (Dol-P). Dol-P is a lipid carrier of the oligosaccharide precursor in N-glycosylation and as a donor of glucose and mannose for several glycosylation pathways1,2,8,12.
Dolichol Biosynthesis
Dolichol is a lipid that can be found in the membrane of the ER and is important for glycosylation in two ways: it acts a lipid carrier for the 14-sugar oligosaccharide that is transferred onto nascent proteins during N-glycosylation and is a monosaccharide donor of mannose and glucose sugars for N-glycosylation, O- and C-mannosylation and GPI anchor biosynthesis (Figure 1)8,13.
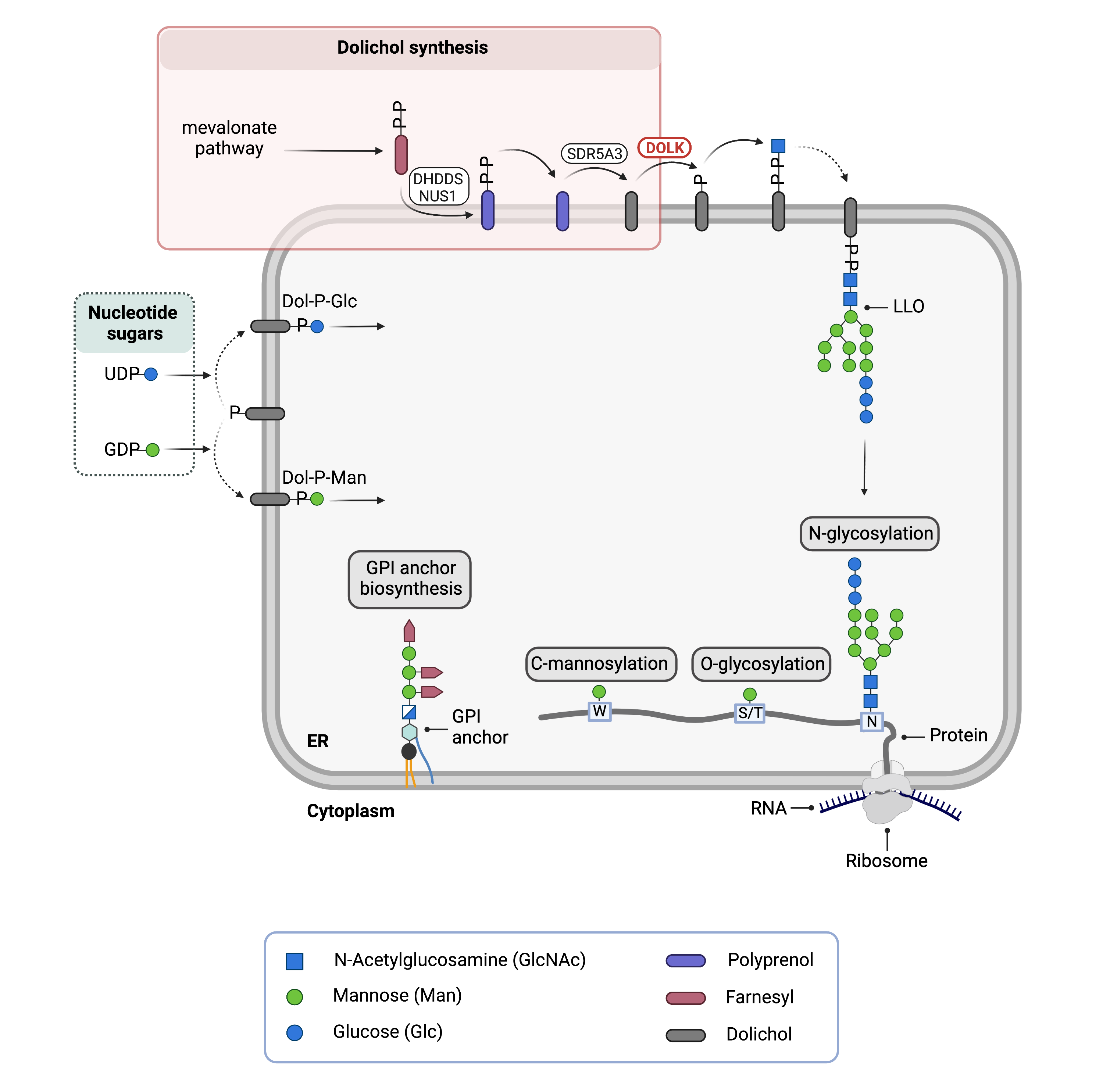
Figure 1.Role of DOLK in glycosylation.
Dolichol kinase (DOLK) catalyzes the phosphorylation of dolichol to generate dolichol-phosphate (Dol-P). In N-glycosylation, the oligosaccharide precursor is built upon Dol-P (together forming the lipid-linked oligosaccharide; LLO) and the 14-sugar oligosaccharide is transferred onto asparagine (N) residues of newly synthesized proteins in the ER lumen. Dol-P is also a donor of glucose (Dol-P-Glc) and mannose (Dol-P-Man) sugars used in multiple glycosylation pathways: N-glycosylation, O-glycosylation and GPI anchor biosynthesis.
Dolichol is synthesised through a series of enzymatic reactions in the cytosol and ER membrane. Once dolichol is generated, it must be phosphorylated by the enzyme dolichol kinase (DOLK), generating dolichol-phosphate (Dol-P) before it can be used as a lipid carrier or monosaccharide donor8,12.
In N-glycosylation, Dol-P acts as a lipid carrier upon which the 14-sugar oligosaccharide, GlcNAc2Man9Glc3, is built. Together, phosphorylated dolichol (Dol-P) and the oligosaccharide form the lipid-linked oligosaccharide (LLO). Once assembly of the LLO is complete, the oligosaccharide component is transferred onto an asparagine residue of a newly synthesized protein the ER.
Sugar donors Dol-P-mannose and Dol-P-glucose are generated upon transfer of mannose and glucose sugars from nucleotide sugars GDP-mannose and UDP-glucose, respectively. These dolichol-linked sugars act as donors of mannose and glucose for multiple glycosylation pathways8,12.
Disease Mechanism
Mutations in the DOLK gene result in an abnormal DOLK enzyme with decreased activity, reducing the transfer of phosphate onto the dolichol and therefore the reducing the amount of phosphate (Dol-P) available for downstream processes8,9,12.
In N-glycosylation, eight Dol-PP molecules are required for the assembly of one N-glycan; reduced availability of Dol-PP is a rate-limiting step in the synthesis of lipid-linked oligosaccharides. Furthermore, as Dol-P is used to generate mannose and glucose sugar donors used in multiple glycosylation pathways, mutations in the DOLK gene can affect multiple body systems and result in severe symptoms1.
Deficient O-mannosylation of alpha-dystroglycan (an adhesion glycoprotein that important for proper functioning of muscle cells) in cardiac muscles due to DOLK deficiency is thought to contribute to dilated cardiomyopathy symptoms2.
Mutations
The DOLK gene is located on Chromosome 9 (9q34.11). 12 pathogenic mutations have been reported in the literature, the majority being missense variants. The missense variant p. Thr520Ala has been reported in unrelated patients and similar dermatopathological changes were observed2,3.
Signs & Symptoms
Individuals with DOLK-CDG typically develop signs and symptoms during infancy into early childhood. DOLK-CDG symptoms are highly variable, and the clinical spectrum has been described to include three major forms; (1) neurological abnormalities with visual impairments and with or without ichthyosis (2) isolated cardiomyopathy or (3) multi-organ involvement with ichthyosis. The clinical presentation of DOLK-CDG may include1–7,11:
- Neurological- developmental delay, poor muscle tone (hypotonia), seizures, microcephaly
- Ophthalmological – progressive nystagmus
- Cardiac – weakened and enlarged heart (dilated cardiomyopathy; sometimes isolated), non-syndromic/asymptomatic dilated cardiomyopathy
- Cutaneous –dry, scaly skin (ichthyosis), reduced hair growth
The most severe symptoms reported include severe congenital ichthyosis and dilated cardiomyopathy in the neonatal period, along with unique phalangeal contractures. Liver and endocrine dysfunctions were also reported, however neurological symptoms were absent2.
Biochemical Presentation
Elevated transaminases, accumulation of iron in the liver, spleen, thymus, thyroid, and pancreas, leading to endocrinological abnormalities, and decreased coagulation factors have been observed in patients3.
Classification
DOLK-CDG is classified as a disorder of multiple glycosylation pathways and within this group, a disorder of dolichol biosynthesis.
Under the former classification system, DOLK-CDG is classified as a Type I CDG, which arise due to defects in the synthesis or transfer of lipid-linked oligosaccharides to N-glycoproteins.
Diagnosis
DOLK-CDG should be considered in individuals presenting with congenital ichthyosis with cardiac involvement and distal digital constrictions in combination with multi-organ failure and coagulation defects. Screening in suspected patients begins with analysis of serum transferrin isoforms with patients often displaying a type I pattern, however, a definitive diagnosis can only be achieved through genetic testing1.
Transferrin Analysis
Affected individuals will typically show a type I pattern by transferrin isoelectric focusing (TIEF)1.
LLO Analysis
No major structural abnormalities in LLO have been detected in patient fibroblasts, however complete chains are missing on transferrin. Reduced amounts of newly synthesized LLO has been detected in patient fibroblasts1.
Biomarkers
No biomarkers specific to DOLK-CDG have been reported.
Prognosis
Due to the broad clinical spectrum of DOLK-CDG, prognosis depends on the severity of an individual’s symptoms.
DOLK-CDG is often fatal, particularly when manifesting with dilated cardiomyopathy. At the most severe end of the clinical spectrum, patients present with lethal neonatal course including severe ichthyosis, digit skin constriction rings, dilated cardiomyopathy, cardiomegaly, hepatosplenomegaly, thrombocytopenia, hypoglycemia, and hypothyroidism1–3,6,7.
At the mild end of the spectrum, 4-month-old and 10-year-old siblings have been reported with DOLK-CDG with only neurological symptoms, which include infantile spasms/seizures, intellectual disability, and autism spectrum disorder5.
Management
Management of DOLK-CDG focuses on managing symptoms dependent on clinical presentation. For cases that do not result in early death, management of symptoms may include a combination of physical therapy, occupational therapy, as well as palliative measures, requiring a multi-disciplinary team.
Therapies
DOLK-CDG patients may have a heart transplant as a potentially curative therapy. Successful transplantation has been preformed on several patients with mild symptoms10,11. Treatment for patients with asymptomatic heart failure and cardiomyopathy include ACE inhibitors and beta-blockers, in addition to diuretics11.
Seizures have been resolved with seizure medications such as vigabatrin for epileptic spasms and carbamazepine for generalized tonic-clonic and focal seizures5.
Research Models
There are several available research models of DOLK-CDG, including yeast and mouse models.
Yeast (S. cerevisiae)
The Sec59 gene is the orthologue of human dolichol kinase (DOLK) and is an essential gene. Temperature sensitive mutants were used to first identify the gene. At the restrictive temperature, Sec59 deficient cells stop dividing, become enlarged, and hypo N-glycosylated secretory proteins accumulate4,14(SGD).
Mouse (M. musculus)
Dolk frameshift mutant mouse
Dolkem1(IMPC)J mutant mice have been generated with a frameshift mutation. Mice homozygous for the mutation are embryonic lethal prior to organ development, surviving to E9.5, or present with preweaning lethality with complete penetrance and survive to early adulthood (IMPC).
Clinical Studies
Active
Active
The Frontiers in Congenital Disorder of Glycosylation Disorders Consortium (FCDGC) is conducting a 5-year natural history study on all CDG types, including DOLK-CDG (NCT NCT04199000). The purpose of this study is to define the natural history and clinical symptoms of CDGs, develop new diagnostic techniques, identify clinical biomarkers that can be used in future clinical trials and evaluate whether dietary treatments improve clinical symptoms and quality of life.
Publications
DOLK-CDG Scientific Articles on PubMed
Additional Resources
DOLK-CDG Infographic
MedlinePlus
IEMbase
OMIM
GARD
NIH
GeneCards
UniProt
Genetic Testing Registry
ClinVar
Marrvel
References
- Kranz, C. et al. A defect in dolichol phosphate biosynthesis causes a new inherited disorder with death in early infancy. American Journal of Human Genetics 80, 433–440 (2007).
- Rush, E. T., Baker, C. v. & Rizzo, W. B. Dolichol kinase deficiency (DOLK-CDG): Two new cases and expansion of phenotype. Am J Med Genet A 173, 2428–2434 (2017).
- Komlosi, K. et al. Fatal Neonatal DOLK-CDG as a Rare Form of Syndromic Ichthyosis. Front Genet 12, 719624. (2021).
- Lefeber, D. J. et al. Autosomal Recessive Dilated Cardiomyopathy due to DOLK Mutations Results from Abnormal Dystroglycan O-Mannosylation. PLoS Genetics 7, 1002427 (2011).
- Helander, A. et al. Dolichol kinase deficiency (DOLK-CDG) with a purely neurological presentation caused by a novel mutation. Mol Genet Metab 110, 342–344 (2013).
- Hall, B. D., Stevenson, R. E. & Jones, J. R. Fatal hyperkeratosis syndrome in four siblings due to dolichol kinase deficiency. Am J Med Genet A 182, 1421–1425 (2020).
- Lieu, M. T. et al. Severe, fatal multisystem manifestations in a patient with dolichol kinase-congenital disorder of glycosylation. Molecular Genetics and Metabolism 110, 484–489 (2013).
- Buczkowska, A., Swiezewska, E. & Lefeber, D. J. Genetic defects in dolichol metabolism. Journal of Inherited Metabolic Disease 38, 157–169 (2015).
- Denecke, J. & Kranz, C. Hypoglycosylation due to dolichol metabolism defects. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1792, 888–895 (2009).
- Klcovansky, J., Mørkrid, L. & Möller, T. Heart transplantation in a child with congenital disorder of glycosylation. J Heart Lung Transplant 35, 1048–1049 (2016).
- Kapusta, L. et al. From discrete dilated cardiomyopathy to successful cardiac transplantation in congenital disorders of glycosylation due to dolichol kinase deficiency (DK1-CDG). Heart Failure Reviews 18, 187–196 (2013).
- Cantagrel, V. & Lefeber, D. J. From glycosylation disorders to dolichol biosynthesis defects: a new class of metabolic diseases. J Inherit Metab Dis 34, 859–867 (2011).
- Welti, M. Regulation of dolichol-linked glycosylation. Glycoconj J 30, 51–56 (2013).
- Fernandez, F., Shridas, P., Jiang, S., Aebi, M. & Waechter, C. J. Expression and characterization of a human cDNA that complements the temperature-sensitive defect in dolichol kinase activity in the yeast sec59-1 mutant: the enzymatic phosphorylation of dolichol and diacylglycerol are catalyzed by separate CTP-mediated kinase activities in Saccharomyces cerevisiae. Glycobiology 12, 555–562 (2002).